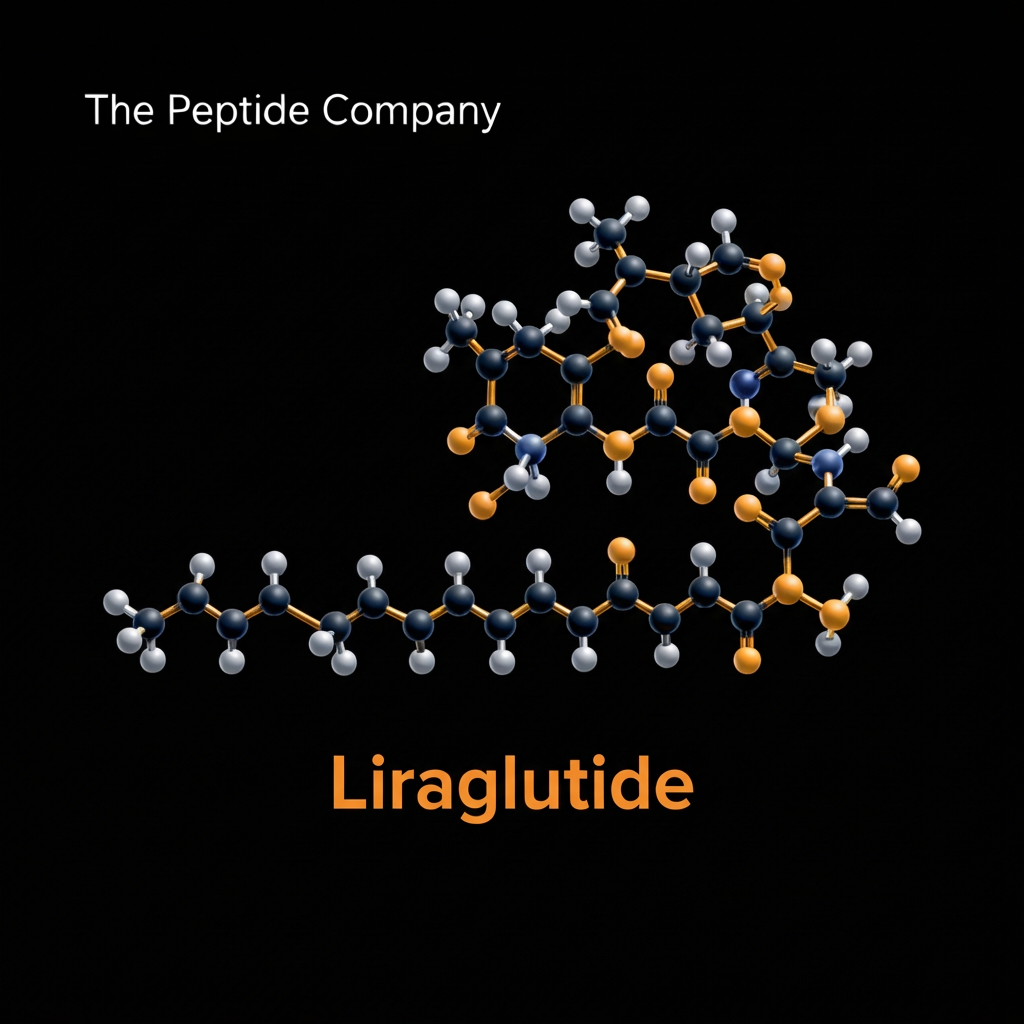
Liraglutide represents a monumental advancement in the field of metabolic peptide engineering, originally developed by research scientists at Novo Nordisk to harness the profound biological potential of the incretin system. The endogenous human incretin hormone, glucagon like peptide 1, is naturally secreted by the intestinal L cells in response to nutrient ingestion. While highly effective at regulating postprandial glucose levels, the native human peptide is rapidly degraded by the enzyme dipeptidyl peptidase 4, resulting in a biological half life of merely two minutes. The development of Liraglutide focused on creating a fatty acid acylated analog capable of resisting this rapid enzymatic destruction while maintaining potent and highly selective receptor activation.
In pharmacological terms, Liraglutide is formally classified as a long acting glucagon like peptide 1 receptor agonist. This classification reflects its ability to mimic the natural hormone and bind to specific G protein coupled receptors located throughout the body. By leveraging advanced lipid attachment technologies, researchers successfully created a molecule that binds to circulating human serum albumin. This clever transport mechanism creates an endogenous reservoir of the peptide within the bloodstream, allowing for a slow, continuous release that provides sustained receptor activation over a twenty four hour period, thereby facilitating once daily dosing in clinical and experimental environments.
When comparing Liraglutide to both the native human hormone and newer generation molecules like semaglutide, significant structural and kinetic differences emerge. While Liraglutide utilizes a sixteen carbon fatty acid chain to achieve its thirteen hour half life, semaglutide utilizes a more complex eighteen carbon diacid chain with a specialized linker, extending its half life to approximately one week. Despite these pharmacokinetic differences, Liraglutide remains a gold standard research compound due to its massive volume of published safety data, its proven efficacy in multiple tissue types, and its highly predictable dose response curve in both rodent and primate models.
Today, the research applications surrounding Liraglutide extend vastly beyond its initial indication for type 2 diabetes mellitus. The scientific community heavily utilizes this peptide to investigate complex physiological networks, including central nervous system pathways governing severe obesity, comprehensive cardiovascular protection models, and progressive neurodegenerative diseases. By evaluating how this single acylated peptide can simultaneously modulate insulin secretion, suppress inflammatory cytokines, and protect vascular endothelium, researchers continue to unlock the profound regenerative capabilities inherent within the mammalian incretin system.
MOLECULAR STRUCTURE AND FATTY ACID ACYLATION CHEMISTRY
The molecular architecture of Liraglutide is a brilliant example of rational peptide design aimed at overcoming the severe pharmacokinetic limitations of native human hormones. The foundation of the Liraglutide molecule maintains a ninety seven percent amino acid sequence homology with the native human glucagon like peptide 1 fragment. To achieve its prolonged biological activity, biochemists engineered two critical structural modifications to the native peptide backbone. The first modification involves a precise amino acid substitution where the natural arginine residue at position 34 is replaced with a lysine residue. This substitution ensures that the subsequent lipid attachment occurs only at the desired location, preventing unwanted structural variations during synthesis. This albumin binding mechanism acts as a slow release biological buffer. Because only a tiny fraction of the Liraglutide dose exists in an unbound, free state at any given moment, the risk of extreme receptor overstimulation is mitigated, resulting in a smooth and predictable pharmacokinetic profile. The spatial arrangement created by the gamma glutamic acid linker ensures that the crucial amino terminal region of the peptide remains fully exposed and geometrically available to interact with the extracellular binding domains of the target receptors.
These precise molecular modifications ensure that Liraglutide retains the exact biological potency of the native hormone while extending its functional half life from a mere two minutes to approximately thirteen hours. This extensive duration of action allows researchers to conduct long term metabolic studies in animal models without the stress and variable baseline fluctuations associated with continuous intravenous infusions or frequent multiple daily injections.
GLP-1 RECEPTOR BINDING AND CAMP SIGNAL TRANSDUCTION
The primary biological effects of Liraglutide are mediated exclusively through its high affinity interaction with the glucagon like peptide 1 receptor, a classic seven transmembrane domain G protein coupled receptor. These highly specialized receptors are densely expressed on the surface of pancreatic beta cells, where they play an absolutely critical role in the regulation of glucose homeostasis. When Liraglutide binds to the extracellular loops of this receptor, it induces a profound conformational change that transfers a mechanical signal across the cellular membrane.
The sudden accumulation of cyclic AMP within the beta cell cytoplasm directly activates two distinct parallel pathways: the protein kinase A pathway and the exchange protein activated by cAMP pathway. Protein kinase A proceeds to phosphorylate numerous downstream targets, including critical voltage dependent calcium channels and ATP sensitive potassium channels. The closure of the potassium channels depolarizes the cellular membrane, allowing a massive influx of extracellular calcium ions. This calcium surge triggers the immediate exocytosis of insulin containing secretory granules.
This multifaceted signal transduction network explains why Liraglutide does not induce severe hypoglycemia in experimental models. By requiring the presence of elevated glucose to trigger insulin release, and by simultaneously suppressing counter regulatory hormones and slowing digestion, the peptide creates a perfectly balanced, multi systemic approach to total metabolic regulation.
BETA CELL PRESERVATION AND PANCREATIC RESEARCH
One of the most intensely researched aspects of Liraglutide involves its profound ability to not only stimulate beta cell function but to actively protect and regenerate the pancreatic islet architecture. Type 2 diabetes is characterized by the progressive failure and death of pancreatic beta cells due to chronic metabolic stress and lipotoxicity. Research models consistently demonstrate that targeted receptor activation by Liraglutide initiates powerful intracellular survival programs that halt this destructive progression.
Following the activation of the primary cyclic AMP cascade, Liraglutide initiates cross talk with the PI3K Akt signaling pathway. The activation of protein kinase B, also known as Akt, acts as a master survival switch within the beta cell. This pathway actively stimulates cellular proliferation and significantly increases the expression of genes responsible for insulin biosynthesis, ensuring the cells have adequate resources to meet metabolic demands.
Furthermore, Liraglutide has been shown to drastically reduce chronic endoplasmic reticulum stress within the pancreatic islets. When beta cells are forced to produce massive quantities of insulin due to peripheral insulin resistance, the endoplasmic reticulum becomes overwhelmed, leading to the accumulation of unfolded proteins and subsequent cellular suicide. The peptide mitigates this stress by enhancing the efficiency of the protein folding machinery and increasing cellular chaperone proteins.
By promoting insulin biosynthesis, protecting against oxidative damage, and directly preventing programmed cell death, Liraglutide offers a comprehensive defensive shield for the pancreas, making it a cornerstone compound in beta cell preservation research.
METABOLIC RESEARCH: WEIGHT REGULATION AND ADIPOSE TISSUE EFFECTS
Beyond its primary role in the pancreas, Liraglutide exerts massive regulatory influence over global energy balance and total body weight. This weight loss mechanism is not simply a secondary side effect of delayed gastric emptying, but rather a profound, direct intervention within the central nervous system. The peptide crosses the blood brain barrier and interacts directly with highly specialized neuronal networks located deep within the hypothalamus.
The specific target for these central effects is the hypothalamic arcuate nucleus, the command center for mammalian appetite regulation. Within this region, Liraglutide heavily stimulates the pro opiomelanocortin neurons, which are responsible for generating powerful signals of satiety and fullness. Simultaneously, the peptide actively suppresses the neuropeptide Y and agouti related peptide neuronal networks, which typically drive hunger and energy conservation behaviors.
The downstream effects of this caloric deficit on adipose tissue are highly favorable for metabolic health. Research data consistently shows that Liraglutide treatment leads to targeted reductions in deep visceral adipose tissue, the highly inflammatory fat depots surrounding the internal organs that heavily contribute to systemic insulin resistance.
By rewiring the neurological drive to consume calories while simultaneously improving the metabolic profile of peripheral fat stores, Liraglutide provides researchers with a highly reliable tool for studying the reversal of severe obesity and its associated metabolic derangements.
CARDIOVASCULAR RESEARCH AND CARDIO PROTECTIVE MECHANISMS
The impact of Liraglutide on cardiovascular health represents one of the most paradigm shifting discoveries in modern peptide pharmacology.
For decades, metabolic treatments focused solely on lowering blood glucose, often failing to reduce the massive cardiovascular mortality rates associated with diabetes. The publication of the landmark LEADER trial shattered this trend, proving that targeted receptor agonism could actively protect the heart and vasculature.
The protective mechanisms are multifaceted and operate both directly and indirectly. The specific receptors for the peptide are heavily expressed directly on cardiac myocytes and the endothelial cells lining the major blood vessels. When Liraglutide activates these vascular receptors, it initiates a powerful anti inflammatory signaling cascade that preserves the integrity of the vascular wall and prevents the initiation of atherosclerosis.
In laboratory models of advanced atherosclerosis, the administration of the peptide actively reduces the formation of dangerous arterial plaques. It achieves this by suppressing the adhesion of circulating monocytes to the vascular wall and preventing their transformation into lipid engorged macrophage foam cells. Furthermore, it actively lowers systemic blood pressure through mild natriuretic effects in the kidneys and direct relaxation of vascular smooth muscle tissue.
These profound cardiovascular benefits have completely rewritten the clinical and experimental approach to metabolic disease, proving that regulating the incretin pathway provides comprehensive protection for the entire mammalian circulatory system.
NEUROPROTECTIVE AND CENTRAL NERVOUS SYSTEM RESEARCH
As researchers recognized the profound ability of Liraglutide to cross the blood brain barrier, investigations rapidly expanded into the realm of advanced neuroprotection. The specific receptors targeted by the peptide are densely expressed in critical cognitive regions, including the hippocampus, the prefrontal cortex, and the basal ganglia. This distribution positions the compound perfectly for interventions in severe neurodegenerative disorders.
In experimental models of Alzheimer’s disease, the brain is characterized by massive insulin resistance, often referred to as type 3 diabetes, alongside the toxic accumulation of amyloid beta plaques. Liraglutide intervenes by restoring normal insulin signaling within the neurons, highly up regulating the production of brain derived neurotrophic factor, and protecting the fragile mitochondrial networks from severe oxidative destruction.
Similar protective effects are observed in Parkinson’s disease research models. The administration of the peptide heavily protects the highly vulnerable dopaminergic neurons residing in the substantia nigra. By suppressing massive neuroinflammation driven by hyperactive microglial cells, the compound prevents the secondary wave of cellular death that typically characterizes progressive movement disorders.
The translation of these neurological findings into clinical reality remains one of the most exciting and intensely pursued avenues of modern peptide science, offering hope for conditions that currently lack any disease modifying treatments.
RENAL AND HEPATIC RESEARCH APPLICATIONS
The systemic benefits of Liraglutide extend deeply into the major metabolic filtration organs, specifically the kidneys and the liver. Diabetic nephropathy is a leading cause of massive renal failure, driven by severe glomerular hyperfiltration, chronic local inflammation, and extreme oxidative stress. The specific receptors targeted by the peptide are highly expressed in the kidney tubular cells, allowing for direct pharmacological intervention.
Research models of severe kidney disease demonstrate that Liraglutide administration significantly reduces total albuminuria, a primary marker of glomerular damage. The peptide exerts profound anti inflammatory renal effects, suppressing the local accumulation of destructive macrophages and preventing the pathological expansion of the mesangial matrix that ultimately chokes the filtration system.
Parallel research in hepatic models heavily focuses on non alcoholic fatty liver disease, a condition characterized by the toxic accumulation of massive lipid droplets within the liver tissue. While the liver lacks dense direct receptor expression, Liraglutide exerts profound indirect benefits by vastly improving global insulin sensitivity, reducing total body weight, and heavily suppressing the flow of toxic free fatty acids from peripheral fat stores to the liver.
These organ specific protective mechanisms demonstrate that incretin modulation is not merely a pancreatic phenomenon, but a massive systemic signaling network that defends the structural integrity of every major metabolic organ in the body.
COMPARATIVE ANALYSIS WITH OTHER GLP-1 RECEPTOR AGONISTS AND TRANSLATIONAL CONSIDERATIONS
Within the rapidly expanding class of incretin based therapies, Liraglutide holds a distinct and highly respected historical position. When conducting a comparative analysis against older first generation agents like exenatide, and newer highly advanced molecules like semaglutide, several critical pharmacological distinctions become immediately apparent.
Exenatide, derived originally from the venom of the Gila monster, features a completely different amino acid sequence and suffers from a highly restricted half life requiring twice daily administration.
Liraglutide solved this issue with its brilliant C16 fatty acid acylation, allowing for smooth once daily dosing. However, the newer generation semaglutide utilizes a much larger C18 diacid chain and a specialized highly stable linker, exponentially increasing albumin binding and allowing for once weekly administration.
Despite the massive successes of this peptide class, significant ongoing research gaps remain. The long term neurological effects of continuous receptor agonism over decades of human use require massive ongoing observation. Furthermore, researchers are actively investigating the potential for combining these peptides with dual agonists targeting the glucose dependent insulinotropic polypeptide or glucagon receptors to drive even more profound metabolic corrections.
As experimental models continue to push the boundaries of peptide science, Liraglutide remains the foundational benchmark against which all future metabolic interventions will be heavily judged, securing its legacy as a true masterpiece of rational molecular engineering.
SOURCED STUDIES
- (1)Knudsen, L. B., et al. “Glucagon-like Peptide-1: The Basis of a New Class of Treatment for Type 2 Diabetes.” JournalofMedicinalChemistry, vol. 43, no. 9, 2000, pp. 1664-1669. DOI: 10.1021/jm9909645.
- (2)Steensgaard, D. B., et al. “The molecular basis for the delayed absorption of the once-daily human GLP-1 analogue, liraglutide.” Diabetes, vol. 57, no. 7, 2008, pp. 1930-1937. DOI: 10.2337/db07-1058.
- (3)Holst, J. J. “The physiology of glucagon-like peptide 1.” PhysiologicalReviews, vol. 87, no. 4, 2007, pp. 1409-1439. DOI: 10.1152/physrev.00034.2006.
- (4)Baggio, L. L., et al. “Biology of incretins: GLP-1 and GIP.” Gastroenterology, vol. 132, no. 6, 2007, pp.2131-2157. DOI: 10.1053/j.gastro.2007.03.054.
- (5)Marso, S. P., et al. “Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes.” NewEngland JournalofMedicine, vol. 375, no. 4, 2016, pp. 311-322. DOI: 10.1056/NEJMoa1603827.
- (6)Astrup, A., et al. “Effects of liraglutide on body weight and on biomarkers of cardiovascular risk in obese subjects.” InternationalJournalofObesity, vol. 33, no. 1, 2009, pp. 1-10. DOI: 10.1038/ijo.2008.212.
- (7)McClean, P. L., et al. “The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease.” JournalofNeuroscience, vol. 31, no. 17, 2011, pp. 6587-6594. DOI: 10.1523/JNEUROSCI.0529-11.2011.
- (8)Armstrong, M. J., et al. “Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN).” TheLancet, vol. 387, no. 10019, 2016, pp. 679-690. DOI: 10.1016/S0140-6736(15)00803-X.
What is Liraglutide primarily studied for?
Liraglutide is primarily studied for its role in GLP-1 receptor activation and its effects on glucose metabolism, insulin signaling, and energy balance in experimental models.
How does Liraglutide interact with the incretin system?
It mimics the activity of endogenous GLP-1, enhancing incretin signaling pathways involved in glucose-dependent insulin release and metabolic regulation.
Why is Liraglutide resistant to rapid degradation?
Liraglutide is structurally modified with a fatty acid side chain, allowing albumin binding and protection from enzymatic breakdown by DPP-4.
What biological pathways are influenced by Liraglutide?
It is studied in pathways related to insulin secretion, glucagon suppression, gastric emptying, appetite signaling, and cardiometabolic regulation.
Is Liraglutide used in cardiometabolic research?
Yes, it is widely studied in experimental models for its association with cardiovascular signaling, inflammation modulation, and metabolic homeostasis.
How does Liraglutide differ from native GLP-1?
Unlike native GLP-1, Liraglutide has an extended half-life due to molecular modifications, enabling prolonged receptor activation and sustained signaling effects.
PMID:
6420373 — Triptorelin and GnRH agonist receptor signaling
6188026 — Continuous GnRH stimulation and pituitary desensitization
6813643 — GnRH agonists and gonadotropin suppression mechanisms
9082563 — Triptorelin effects on LH and FSH secretion
1905666 — GnRH analog modulation of pituitary signaling
1924466 — Hypothalamic-pituitary-gonadal axis suppression research
2145588 — Pharmacology of Triptorelin and GnRH analogs
8390782 — Long-acting GnRH agonist endocrine modulation
10446354 — GnRH receptor downregulation mechanisms
16886967 — Triptorelin and reproductive hormone regulation
RELATED SEARCHES:
Semaglutide : GLP-1 Receptor Agonism, Incretin Signaling, and Metabolic Regulation
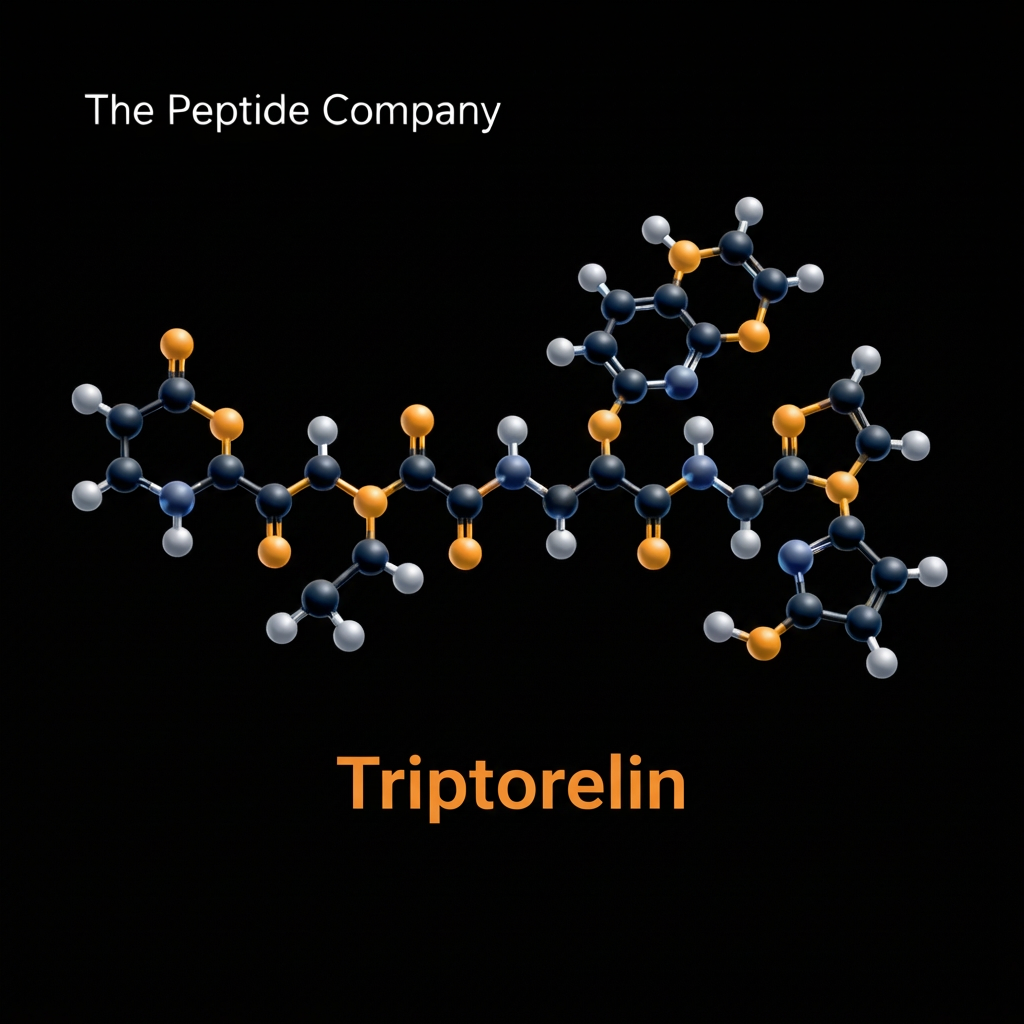
Triptorelin is a highly potent synthetic decapeptide analog of the endogenous gonadotropin releasing hormone naturally produced within the hypothalamus. Developed through extensive biochemical engineering to manipulate the complex feedback loops of the human endocrine system, this peptide compound has become a foundational tool in advanced reproductive and oncological research. The primary objective behind the creation of Triptorelin was to design a molecule capable of safely and reversibly regulating the synthesis of key reproductive hormones by targeting the anterior pituitary gland directly. This targeted approach allows researchers to study profound hormonal suppression without the need for irreversible surgical interventions in experimental models.
As a specific gonadotropin releasing hormone agonist, Triptorelin exhibits a highly unique pharmacological behavior characterized by a paradoxical desensitization mechanism. Under normal physiological conditions, endogenous gonadotropin releasing hormone is secreted in a pulsatile manner, which stimulates the pituitary gland to release luteinizing hormone and follicle stimulating hormone. When Triptorelin is introduced in a continuous, non pulsatile formulation, it initially acts as a superagonist, causing a massive initial release of these pituitary hormones. However, sustained exposure to the synthetic peptide rapidly overstimulates the receptors, leading to their internal cellular degradation and a subsequent complete shutdown of gonadotropin production.
This biphasic response is central to the wide array of experimental research applications currently utilizing Triptorelin. By intentionally crashing the production of luteinizing hormone and follicle stimulating hormone, researchers can effectively eliminate the downstream synthesis of gonadal steroids, namely testosterone in males and estradiol in females. This profound state of biochemical castration or medically induced menopause provides an ideal physiological environment for studying hormone dependent conditions. Laboratory models frequently leverage this mechanism to investigate the progression and regression of steroid sensitive pathologies over extended timeframes.
Today, the research landscape surrounding Triptorelin encompasses diverse medical disciplines ranging from reproductive endocrinology to advanced neurobiology. Primary investigative areas include the suppression of hormone dependent tumor models, such as advanced prostate and breast cancer cell lines, as well as the management of severe endometriosis and uterine fibroids in female animal models.
Furthermore, emerging research is actively exploring the secondary systemic effects of profound sex steroid deprivation, including changes in bone mineral density, cognitive function, and cardiovascular health, making Triptorelin a molecule of immense translational value in modern peptide science.
MOLECULAR STRUCTURE AND GNRH ANALOG CHEMISTRY
The molecular architecture of Triptorelin is a masterclass in rational peptide design. The endogenous gonadotropin releasing hormone is a decapeptide characterized by the amino acid sequence pyroglutamate histidine tryptophan serine tyrosine glycine leucine arginine proline glycine amide. While highly effective in its natural biological context, this native peptide has an extremely short biological half life of approximately two to four minutes due to rapid enzymatic degradation by specific endopeptidases located in the hypothalamus and pituitary tissues. To overcome this limitation for research purposes, biochemists modified the native sequence to enhance both stability and receptor affinity.
The specific substitution of D Tryptophan at position six does more than just protect the peptide from enzymatic breakdown. This structural adjustment significantly increases the binding affinity of Triptorelin for the gonadotropin releasing hormone receptor located on the surface of pituitary gonadotroph cells. Research indicates that the binding affinity of Triptorelin is approximately one hundred times greater than that of the native peptide. This immense receptor affinity ensures that the synthetic analog outcompetes endogenous signaling molecules, maintaining dominant control over the receptor complex even at relatively low circulating concentrations.
The development of these advanced depot formulations revolutionized the use of Triptorelin in laboratory environments. By engineering a delivery matrix that slowly degrades via passive hydrolysis in the tissue, researchers can maintain continuous receptor saturation without the need for daily subcutaneous injections. This sustained release chemistry is the ultimate driver of the paradoxical desensitization effect, as the pituitary gland is never granted a recovery window to upregulate new receptor proteins.
GNRH RECEPTOR BINDING AND PITUITARY DESENSITIZATION MECHANISMS
The primary target of Triptorelin is the gonadotropin releasing hormone receptor, a classic seven transmembrane G protein coupled receptor expressed predominantly on the surface of gonadotroph cells within the anterior pituitary gland. The sequence of cellular events that follows Triptorelin binding is highly complex and illustrates the intricate feedback mechanisms inherent to endocrine cells. Initially, the binding of the superagonist triggers a robust classical signaling cascade that rapidly upregulates hormone secretion.
This initial flare effect is a critical consideration in experimental research, as it temporarily exacerbates the very hormonal environment the peptide is designed to suppress. Following this initial period of hyperstimulation, the continuous presence of Triptorelin forces the cellular machinery into a defensive posture. The constant activation of the intracellular signaling cascades triggers complex negative feedback loops designed to protect the cell from toxic overstimulation.
Through this elegant mechanism of induced cellular exhaustion, Triptorelin effectively silences the pituitary gland. The uncoupling of the Gq protein cascade ensures that even if trace amounts of functional receptors remain on the cell surface, they cannot transmit the signal required to manufacture new hormones. This state of profound desensitization is entirely reversible; once the continuous delivery of the peptide ceases and the remaining molecules are cleared from the system, the pituitary gradually synthesizes new receptor proteins and restores normal pulsatile function.
HYPOTHALAMIC PITUITARY GONADAL AXIS SUPPRESSION
The ultimate systemic consequence of pituitary desensitization is the complete suppression of the hypothalamic pituitary gonadal axis. This physiological axis relies on a delicate balance of positive and negative feedback loops to maintain normal reproductive function. The hypothalamus releases gonadotropin releasing hormone to stimulate the pituitary, which releases luteinizing hormone and follicle stimulating hormone to stimulate the gonads. The gonads, in turn, produce sex steroids that feed back to the brain to modulate further hormone release. Triptorelin completely severs this communication chain at the pituitary level.
This biochemical castration is remarkably consistent and highly reproducible across diverse mammalian species, making it an invaluable standard in laboratory research. In female models, the suppression of follicle stimulating hormone prevents the maturation of ovarian follicles, while the lack of luteinizing hormone halts the production of estradiol and progesterone. This effectively induces a state of profound hypoestrogenism, simulating a complete menopausal transition.
The disruption of the feedback loop also affects higher regulatory centers in the brain. Because the gonads are no longer producing steroids, the hypothalamus perceives a severe hormonal deficit and attempts to compensate by upregulating its own production of endogenous gonadotropin releasing hormone. However, because the pituitary receptors remain blocked and degraded by the continuous presence of Triptorelin, these hypothalamic efforts are completely futile. This isolated hypothalamic activity provides researchers with a unique window into the independent functioning of distinct brain regions during states of profound systemic hormonal deprivation.
RESEARCH APPLICATIONS IN HORMONE DEPENDENT TUMOR MODELS
The most prominent application of Triptorelin in modern biomedical research involves the investigation of hormone dependent oncology models. Many abnormal cellular proliferations, particularly those originating in reproductive tissues, rely heavily on circulating androgens or estrogens to fuel their rapid division and prevent cellular apoptosis. By utilizing Triptorelin to eliminate these fuel sources, researchers can meticulously study the mechanisms of tumor growth arrest and cellular death.
While the initial regression of tumor volume is significant, research models also utilize Triptorelin to study the inevitable development of castration resistant disease states. Over prolonged periods of complete androgen deprivation, certain cancer cell lines mutate to synthesize their own localized androgens or develop hypersensitive receptors that activate in the absence of traditional ligands. Triptorelin provides the necessary baseline suppression required to observe and map these complex cellular escape mechanisms.
Similarly, in female research models, Triptorelin is utilized to investigate estrogen dependent conditions including specific phenotypes of breast cancer, advanced endometriosis, and large uterine fibroids. By completely suppressing ovarian estradiol production, researchers can evaluate the regression of ectopic endometrial tissue implants and monitor the shrinkage of fibroid masses. The peptide allows for the careful study of angiogenesis inhibition within these abnormal tissues, as the lack of estrogen signaling significantly reduces the expression of vascular endothelial growth factor, essentially starving the abnormal growths of their local blood supply.
REPRODUCTIVE BIOLOGY AND FERTILITY RESEARCH
In a fascinating paradox, while Triptorelin is primarily known for suppressing the reproductive axis, it is also a cornerstone compound in the research and development of advanced fertility treatments. In the context of controlled ovarian stimulation protocols utilized in in vitro fertilization research, achieving absolute control over the hormonal environment is critical to ensure the simultaneous maturation of multiple viable oocytes.
Beyond female fertility models, Triptorelin is actively investigated in male reproductive biology, particularly regarding spermatogenesis and potential contraceptive applications. Deep suppression of luteinizing hormone and follicle stimulating hormone severely impairs the function of the seminiferous tubules, leading to a massive reduction in sperm count and motility. Researchers carefully monitor the time course of this suppression and the subsequent recovery phase after peptide withdrawal to evaluate the feasibility and safety of temporary biochemical sterilization in mammalian subjects.
The absolute reversibility of Triptorelin induced suppression remains a major focus of fertility preservation research. Experimental protocols often utilize the peptide to temporarily shut down the reproductive axis in juvenile models prior to the administration of highly toxic chemotherapy agents. Researchers hypothesize that putting the delicate gonadal stem cells into a dormant, metabolically inactive state may protect them from the cytotoxic damage typically caused by harsh alkylating agents, thereby preserving long term reproductive potential following cancer treatments.
NEURO PROTECTIVE AND CENTRAL NERVOUS SYSTEM RESEARCH
While the peripheral effects of Triptorelin on the gonads are well mapped, emerging research is rapidly expanding into the central nervous system. Modern immunohistochemical mapping has identified the unexpected presence of specific gonadotropin releasing hormone receptors across various regions of the brain, most notably within the cerebral cortex and the hippocampus, areas intimately associated with memory consolidation and complex executive function.
This discovery has launched novel investigations into the potential neuroprotective effects of manipulating these central receptors.
Researchers are exploring how the direct binding of Triptorelin to hippocampal neurons might influence the production of local neurotrophic factors independently of the systemic suppression of sex steroids. Additionally, the profound systemic loss of estrogen and testosterone induced by the peptide presents a unique model for studying the cognitive consequences of sudden hormone deprivation.
These intricate models highlight the double edged nature of profound hormonal manipulation. While researchers carefully document potential declines in spatial memory and psychomotor speed following the removal of systemic testosterone and estrogen, they simultaneously investigate whether the direct central action of the peptide can mitigate these effects. This ongoing research is critical for understanding the long term neurological safety profile of prolonged hormone suppression therapies.
BONEMINERAL DENSITY ANDMETABOLIC RESEARCH IMPLICATIONS
A massive secondary area of scientific inquiry regarding Triptorelin centers on the profound metabolic consequences of long term sex steroid deprivation, most specifically the rapid acceleration of bone remodeling and subsequent loss of bone mineral density. Both estrogen and testosterone play mandatory, continuous roles in maintaining the structural integrity of the mammalian skeleton by regulating the delicate balance between bone forming osteoblasts and bone resorbing osteoclasts.
When Triptorelin drives sex steroids down to castrate or menopausal levels, the physiological brakes on bone resorption are completely removed. This creates a highly accelerated, high turnover state within the skeletal matrix. Researchers utilize this predictable mechanism to create highly accurate animal models of severe osteoporosis and advanced osteopenia in relatively short timeframes, allowing for the rapid testing of novel bone targeted therapeutic agents.
To combat these severe metabolic consequences in long term study designs, researchers frequently employ add back therapy frameworks. This involves the continuous administration of Triptorelin to maintain total suppression of the endogenous gonadal axis, coupled with the highly controlled, low dose reintroduction of specific synthetic estrogens or progestins. This allows scientists to determine the absolute minimum threshold of steroid hormones required to maintain bone health and cardiovascular lipid profiles without stimulating the primary hormone dependent tumor models under investigation.
COMPARATIVE ANALYSIS AND TRANSLATIONAL RESEARCH CONSIDERATIONS
Within the highly specialized landscape of endocrine modulation, Triptorelin is frequently compared against other prominent analogs such as leuprolide and goserelin. While all these peptides operate via the exact same receptor downregulation mechanism, subtle differences in their engineered amino acid sequences dictate variations in their precise binding affinities, local tissue distribution, and compatibility with different sustained release polymer matrices. These minor pharmacokinetic variations allow researchers to select specific analogs based on the desired duration and depth of suppression required for particular experimental protocols.
More recently, translational research has focused heavily on comparing Triptorelin and other traditional agonists against the newer class of competitive gonadotropin releasing hormone antagonists, such as degarelix. Unlike Triptorelin, which requires weeks of receptor overstimulation and internalization to achieve suppression, antagonists simply block the receptor binding site immediately, achieving castrate levels of hormones within twenty four to forty eight hours without any initial stimulatory flare effect.
Moving forward, research gaps remain regarding the ultimate long term cellular toxicity of sustained receptor internalizations and the full scope of extra pituitary receptor activation in the brain and immune system. As peptide synthesis technology continues to evolve, the extensive data gathered from decades of Triptorelin research will undoubtedly inform the development of next generation, highly targeted neuroendocrine modulators designed to manipulate specific cellular pathways with unprecedented precision.
SOURCEDSTUDIES
- (1)Schally, A. V., et al. “Luteinizing hormone releasing hormone and its analogues: Recent basic and clinical investigations.” InternationalJournalofGynaecologyandObstetrics, vol. 18, no. 5, 1980, pp. 318-324. DOI: 10.1002/j.1879-3479.1980.tb00293.x.
- (2)Conn, P. M., et al. “Gonadotropin releasing hormone and its analogues.” TheNewEnglandJournal ofMedicine, vol. 324, no. 2, 1991, pp. 93-103. DOI: 10.1056/NEJM199101103240205.
- (3)Huirne, J. A., et al. “Gonadotropin releasing hormone receptor agonists and antagonists.” The Lancet, vol. 358, no. 9295, 2001, pp. 1793-1803. DOI: 10.1016/S0140-6736(01)06806-0.
- (4)Belchetz, P. E., et al. “Hypophysial responses to continuous and intermittent delivery of hypothalamic gonadotropin releasing hormone.” Science, vol. 202, no. 4368, 1978, pp. 631-633. DOI: 10.1126/science.100883.
- (5)Seidenfeld, J., et al. “Single therapy androgen suppression in men with advanced prostate cancer: A systematic review and meta analysis.” AnnalsofInternalMedicine, vol. 132, no. 7, 2000, pp. 566-577. DOI: 10.7326/0003-4819-132-7-200004040-00009.
- (6)Huggins, C., et al. “Studies on prostatic cancer: The effects of castration on advanced carcinoma of the prostate gland.” ArchivesofSurgery, vol. 43, no. 2, 1941, pp. 209-223. DOI: 10.1001/archsurg.1941.01210140043004.
- (7)Maclon, C. B., et al. “The history of the in vitro fertilization cycle: A focus on the role of gonadotropin releasing hormone agonists.” HumanReproductionUpdate, vol. 12, no. 4, 2006, pp. 411-421. DOI: 10.1093/humupd/dml016.
- (8)Casadesus, G., et al. “Modulation of amyloid beta precursor protein processing by luteinizing hormone: A highly relevant pathway in Alzheimer disease.” JournalofNeurochemistry, vol. 97, no. 5, 2006, pp. 1309-1315. DOI: 10.1111/j.1471-4159.2006.03823.x.
FAQ:
What is Triptorelin?
Triptorelin is a synthetic decapeptide analog of gonadotropin-releasing hormone (GnRH) studied for its effects on hypothalamic-pituitary-gonadal axis signaling.
How does Triptorelin interact with GnRH receptors?
Triptorelin binds to GnRH receptors in the anterior pituitary, initially stimulating gonadotropin release followed by receptor desensitization with sustained exposure.
What happens after prolonged Triptorelin signaling?
Continuous receptor activation leads to downregulation of pituitary GnRH receptors and suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) signaling.
What biological axis is affected by Triptorelin?
Triptorelin is primarily studied for its modulation of the hypothalamic-pituitary-gonadal (HPG) axis.
Is Triptorelin a GnRH agonist or antagonist?
Triptorelin is classified as a GnRH agonist that produces functional suppression through receptor desensitization over time.
Why is Triptorelin used in endocrine research models?
It is utilized to study gonadotropin suppression, hormonal feedback loops, and reproductive axis signaling.
Does Triptorelin initially increase hormone signaling?
Yes, initial GnRH receptor stimulation can transiently increase LH and FSH release before downregulation occurs.
How many amino acids are in Triptorelin?
Triptorelin is a decapeptide composed of ten amino acids.
What makes Triptorelin different from native GnRH?
Triptorelin is structurally modified to enhance receptor affinity and prolong biological signaling compared to endogenous GnRH.
PMID:
PMID: 6420373 — Triptorelin and GnRH agonist receptor signaling
PMID: 6138026 — Continuous GnRH stimulation and pituitary desensitization
PMID: 6813643 — GnRH agonists and gonadotropin suppression mechanisms
PMID: 2982563 — Triptorelin effects on LH and FSH secretion
PMID: 1905466 — GnRH analog modulation of pituitary signaling
PMID: 1924436 — Hypothalamic pituitary gonadal axis suppression research
PMID: 2145588 — Pharmacology of Triptorelin and GnRH analogs
PMID: 8390782 — Long-acting GnRH agonist endocrine modulation
PMID: 10443654 — GnRH receptor downregulation mechanisms
PMID: 16886967 — Triptorelin and reproductive hormone regulation
RELATED SEARCHES:
Activin A: TGF-β Superfamily Signaling, SMAD2/3 Pathway Regulation, and Muscle–Fibrosis Cross-Talk
Myostatin (GDF-8): Muscle Growth Regulation, TGF-β Superfamily Signaling, and Anabolic Homeostasis
Semaglutide : GLP-1 Receptor Agonism, Incretin Signaling, and Metabolic Regulation
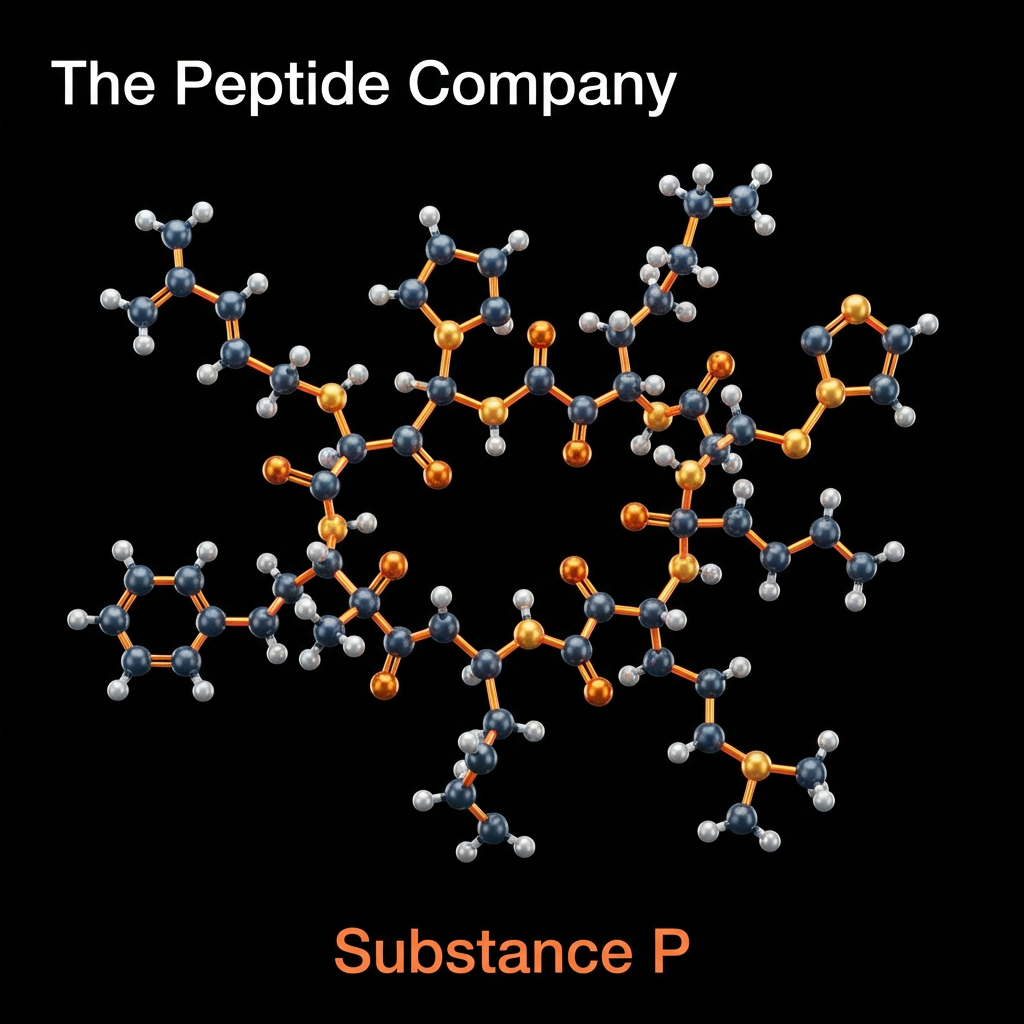
Substance P (SP) is an 11-amino acid neuropeptide belonging to the tachykinin family, widely distributed throughout the central and peripheral nervous systems. First identified in 1931 by von Euler and Gaddum, it holds the distinction of being the first neuropeptide to be discovered. In contemporary research, Substance P is extensively studied as a primary mediator of nociception (pain transmission) and neurogenic inflammation. It functions principally through high-affinity binding to the Neurokinin-1 (NK1) receptor, a G-protein coupled receptor (GPCR) found on neurons, immune cells, and endothelial tissues.
The peptide sequence of Substance P (Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-NH2) is highly conserved across mammalian species, reflecting its critical evolutionary role in survival mechanisms. Beyond its classical function as a neurotransmitter of pain signals in the dorsal horn of the spinal cord, Substance P is now recognized as a ubiquitous modulator of physiological stress responses, immune system regulation, and emotional behavior. Research models involving NK1 receptor antagonism have provided profound insights into the peptide’s involvement in conditions ranging from chronic pain and inflammatory diseases to depression and anxiety disorders.
MOLECULAR STRUCTURE AND NEURO KININ RECEPTOR FAMILY
Substance P is encoded by the preprotachykinin-A (TAC1) gene and is synthesized as a larger precursor protein that undergoes post-translational cleavage. It belongs to the tachykinin peptide family, which also includes Neurokinin A and Neurokinin B. These peptides share a common C-terminal sequence, Phe-X-Gly-Leu-Met-NH2, which is essential for receptor activation. However, Substance P exhibits unique selectivity for the NK1 receptor, distinguishing its biological profile from other family members that prefer NK2 or NK3 receptors.
Structural biology research has elucidated that the C-terminal region of Substance P is responsible for receptor binding and activation, while the N-terminal sequence determines metabolic stability and specificity. The amidation of the C-terminus is critical for biological potency; non-amidated analogs show drastically reduced affinity for the NK1 receptor. This structure-activity relationship is a key focus in the development of synthetic NK1 antagonists designed to block Substance P signaling without interfering with other tachykinin pathways.
The most well-characterized role of Substance P is its function as a nociceptive neurotransmitter. It is synthesized in the cell bodies of dorsal root ganglion (DRG) neurons and transported to both central and peripheral nerve terminals. In response to noxious stimuli—such as intense heat, mechanical pressure, or chemical irritants—Substance P is released into the dorsal horn of the spinal cord, where it acts synergistically with glutamate to transmit pain signals to the brain.
Research into chronic pain models has demonstrated that inhibiting Substance P signaling can attenuate central sensitization, the process by which the nervous system becomes hypersensitive to stimuli. This has led to extensive investigation of NK1 receptor antagonists as potential analgesics. Although early clinical trials faced challenges due to species-specific differences in receptor pharmacology, preclinical data continues to support the critical role of Substance P in the maintenance of neuropathic and inflammatory pain.
NEUROIMMUNE INTERACTIONS AND INFLAMMATORY REGULATION
Substance P serves as a vital bridge between the nervous and immune systems. NK1 receptors are expressed on a wide array of immune cells, including macrophages, mast cells, T-lymphocytes, and dendritic cells. The release of Substance P from peripheral nerve endings can directly activate these cells, triggering the release of pro-inflammatory cytokines like IL-1β, IL-6, and TNF-α. This bi-directional communication is fundamental to the concept of neurogenic inflammation.
This neuroimmune axis is particularly relevant in research on stress-induced immune suppression and inflammatory disorders. By modulating cytokine profiles, Substance P can influence the progression of inflammatory responses. Experimental models have shown that NK1 receptor blockade can reduce the severity of inflammation in conditions like colitis and arthritis, highlighting the therapeutic potential of targeting this neuropeptide in autoimmune pathology.
NEUROGENIC INFLAMMATION AND WOUND HEALING RESEARCH
Neurogenic inflammation refers to the process where inflammatory symptoms—redness, swelling, and heat—are initiated by the release of neuropeptides from sensory nerves rather than by immune cells directly. Substance P is the primary mediator of this process. Upon release from peripheral nerve terminals, it causes vasodilation and plasma protein extravasation (leakage of fluid from blood vessels) by acting on endothelial cells.
Research in regenerative medicine is exploring the dual nature of Substance P. While its pro-inflammatory effects can be detrimental in chronic disease, its ability to stimulate cellular proliferation and angiogenesis is beneficial for tissue repair. Investigational therapies aim to harness its wound-healing properties—particularly in neuropathic ulcers and corneal injuries—while managing the inflammatory component.
SUBSTANCE P IN NEUROPSYCHIATRIC AND STRESS RESPONSE MODELS
Beyond the spinal cord and periphery, Substance P is highly concentrated in brain regions associated with emotional processing, such as the amygdala, hypothalamus, and hippocampus. It is a key component of the stress response system. Animal models of stress and anxiety consistently show elevated release of Substance P in these limbic areas, suggesting it functions as a “stress neurotransmitter.”
Although clinical translation for depression has been mixed, research
continues to investigate Substance P antagonists for specific stress-related disorders, including PTSD and alcohol dependence. The hypothesis is that blocking NK1 signaling may dampen the overactive stress circuitry without the sedative side effects common to benzodiazepines.
PHARMACOLOGICAL MODULATION: NK1 RECEPTOR ANTAGONISTS
The broad physiological role of Substance P has driven the development of NK1 receptor antagonists. The most successful clinical application to date is aprepitant, used to prevent chemotherapy-induced nausea and vomiting (CINV). Substance P is a major mediator of the vomiting reflex in the brainstem, and blocking its receptor effectively suppresses this response.
SOURCEDSTUDIES
- (1)Steinhoff, M.S., et al. “The role of neurokinin-1 receptors in physiology and pathophysiology.” PhysiologicalReviews, vol. 94, no. 1, 2014, pp. 265-301. DOI: 10.1152/physrev.00040.2012.
- (2)Zieglgänsberger, W. “Substance P and pain chronicity.” CellandTissueResearch, vol. 375, no. 1, 2019, pp. 227-241. DOI: 10.1007/s00441-018-2922-y.
- (3)Mashaghi, A., et al. “Neuropeptide substance P and the immune response.” CellularandMolecular LifeSciences, vol. 73, no. 22, 2016, pp. 4249-4264. DOI: 10.1007/s00018-016-2293-z.
- (4)Suvas, S. “Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis.” Journal ofImmunology, vol. 199, no. 5, 2017, pp. 1543-1552. DOI: 10.4049/jimmunol.1601751.
- (5)Ebner, K., et al. “Substance P in stress and anxiety: behavioral changes, mechanisms, and novel therapeutic targets.” CurrentOpinioninPharmacology, vol. 6, no. 5, 2006, pp. 475-481. DOI: 10.1016/j.coph.2006.05.004.
- (6)Navari, R.M., et al. “Neurokinin-1 receptor antagonists in the treatment of chemotherapy-induced nausea and vomiting.” DrugDesign,DevelopmentandTherapy, vol. 10, 2016, pp. 2567-2575. DOI: 10.2147/DDDT.S109404.
FAQ:
What is Substance P?
Substance P is an 11-amino acid neuropeptide belonging to the tachykinin family, widely distributed throughout the central and peripheral nervous systems.
What receptor does Substance P primarily interact with?
Substance P primarily binds to the neurokinin-1 (NK1) receptor, a G-protein coupled receptor expressed on neurons, immune cells, and endothelial tissues.
What biological processes is Substance P associated with?
It is studied for its role in nociceptive transmission, neurogenic inflammation, neuroimmune signaling, and sensory neuron communication.
Where is Substance P found in the body?
Substance P is present in the brain, spinal cord, peripheral sensory neurons, and various immune-related tissues.
How does Substance P relate to pain signaling?
It is widely researched as a neurotransmitter involved in transmitting nociceptive signals between peripheral nerves and central nervous system pathways.
Is Substance P involved in inflammation pathways?
Research indicates Substance P participates in neurogenic inflammation through interactions with immune cells and vascular signaling.
What type of peptide is Substance P?
Substance P is a tachykinin neuropeptide composed of 11 amino acids with conserved structure across mammalian species.
Why is Substance P studied in neuroimmune research?
It is investigated for its role in communication between the nervous system and immune signaling pathways.
Does Substance P act in both central and peripheral systems?
Yes, Substance P is studied in both central nervous system signaling and peripheral sensory neuron pathways.
PMID:
PMID: 7681072 — Distribution and function of Substance P in the nervous system
PMID: 7542593 — Neurokinin-1 receptor binding and Substance P signaling
PMID: 1374954 — Substance P and nociceptive transmission mechanisms
PMID: 1714860 — Tachykinins and neurogenic inflammation pathways
PMID: 7521983 — Substance P in neuroimmune communication
PMID: 1693883 — Sensory neuron release of Substance P
PMID: 8381513 — NK1 receptor pharmacology and signaling
PMID: 10844083 — Substance P in central nervous system signaling
PMID: 11877335 — Substance P and inflammatory mediator pathways
PMID: 15265877 — Neurokinin receptors and peptide neurotransmission
RELATED SEARCHES:
Cerebrolysin : Neurotrophic Peptide
Noopept : Neuropeptide Derived
Thymalin: Thymic Bioregulator Peptide, Immune Aging, and Epigenetic Control of Cellular Homeostasis
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research

MixtureResearch, BDNFandNGF Signaling, and Neuroprotective Mechanisms in Neurological Models
Cerebrolysin represents a highly sophisticated and extensively researched neurotrophic peptide mixture utilized in advanced neurological recovery models. The historical development of this unique compound traces back several decades, originating from an intricate process of extracting and purifying brain tissue. Specifically, the formulation is derived from purified porcine brain proteins, which are carefully processed to create a stable, biologically active therapeutic agent. Over the years, the scientific community has heavily scrutinized this preparation, transitioning its status from a traditional biological extract to a highly defined, multi component polypeptide bioregulator capable of exerting profound disease modifying effects within the central nervous system.
In pharmacological terms, Cerebrolysin is classified as a complex peptide mixture rather than a single synthetic molecule. This classification is vital to understanding its broad spectrum mechanism of action. Unlike modern synthetic drugs that typically target a single receptor or isolated enzymatic pathway, this biological preparation mimics the highly complex, synergistic signaling environments naturally found within a healthy mammalian brain. The diverse array of small peptides operates collectively to modulate entire networks of cellular survival, structural remodeling, and metabolic efficiency, establishing a pharmacological profile that is notoriously difficult to replicate with isolated synthetic compounds.
The manufacturing process of Cerebrolysin relies on standardized enzymatic hydrolysis, a highly controlled biochemical technique designed to break down massive, immunogenic porcine brain proteins into tiny, safe, and biologically active fragments. By utilizing specific proteolytic enzymes under strict laboratory conditions, researchers ensure that the resulting peptide mixture is devoid of large proteins, lipids, and antigenic cellular debris. This meticulous enzymatic cleavage
generates a consistent pool of low molecular weight peptides that can
safely navigate systemic circulation and ultimately penetrate the highly restrictive protective barriers of the human brain.
Today, the neurotrophic and neuroprotective research applications surrounding Cerebrolysin are vast and encompass some of the most challenging conditions in modern neurology. Experimental models heavily utilize this peptide complex to investigate neuronal survival following acute ischemic stroke, progressive neurodegeneration in Alzheimer’s disease, diffuse axonal injury in severe traumatic brain injury, and chronic cognitive decline associated with vascular dementia. By evaluating how this mixture influences the brain at a fundamental molecular level, scientists continue to uncover the profound regenerative capabilities of natural neurotrophic signaling.
COMPOSITIONANDMOLECULARCHARACTERISTICS
The biochemical composition of Cerebrolysin is highly distinctive and serves as the foundation for its pleiotropic therapeutic effects. The rigorous enzymatic hydrolysis process yields a final solution characterized primarily by an abundance of very low molecular weight peptides. Analytical laboratory techniques consistently confirm that the vast majority of the active peptide fragments within the formulation are smaller than 10 kilodaltons. This specific molecular weight distribution is an absolute prerequisite for central nervous system research, as larger protein molecules are categorically excluded from entering the brain parenchyma.
The free amino acid content, which constitutes a significant portion of the total dry weight, includes vital neuroactive building blocks such as glutamate, aspartate, and glycine. However, the primary pharmacological
activity is attributed to the active neuropeptide fractions. These small
peptide sequences act as direct molecular mimics of naturally occurring human survival factors. By maintaining such a low molecular weight profile, the active constituents easily bypass the formidable obstacles that typically hinder neurological drug delivery, ensuring that the active signaling molecules reach their intended targets within the cerebral cortex and hippocampus.
This molecular weight distribution distinctly separates Cerebrolysin from synthetic single peptides. While a synthetic peptide contains only one specific amino acid sequence designed for one specific target, the complex mixture approach ensures that multiple cellular receptors are activated simultaneously. This multi targeted biological strategy effectively prevents the cellular desensitization and feedback inhibition frequently observed in single molecule experimental pharmacology.
BDNF,NGF,CNTF,ANDNT-3MIMETICACTIVITY
The most extensively researched aspect of Cerebrolysin is its ability to directly mimic the action of multiple endogenous neurotrophic factors simultaneously. Neurotrophins are specialized proteins that regulate the growth, survival, and functional maintenance of neurons. By presenting a complex matrix of peptide analogs, Cerebrolysin essentially functions as a broad spectrum neurotrophic substitute, capable of stimulating numerous survival pathways that are typically compromised during severe neurological disease or acute brain injury.
In addition to mimicking brain derived neurotrophic factor, researchers have documented profound nerve growth factor mimetic effects. Nerve growth factor is absolutely critical for the survival of cholinergic neurons, which are heavily implicated in memory formation and cognitive processing. Cerebrolysin contains peptide sequences that successfully bind and activate the TrkA receptor, replicating the natural survival signals required by these vulnerable cholinergic networks. This specific interaction is a primary focus in laboratory models of progressive memory loss.
This simultaneous, synergistic activation is why the peptide mixture is so heavily favored in advanced research. The brain relies on a highly coordinated symphony of growth factors to maintain synaptic health. By delivering a mixture that engages TrkA, TrkB, and other neurotrophic receptors concurrently, researchers can successfully replicate the complex biological conditions necessary for profound tissue regeneration and neurological repair.
NEUROPROTECTIONINISCHEMICSTROKEMODELS
During an acute ischemic stroke, the sudden interruption of cerebral blood flow deprives brain tissue of oxygen and glucose, initiating a catastrophic biochemical cascade known as glutamate excitotoxicity. As energy reserves deplete, dying neurons release massive quantities of glutamate, which hyperactivates neighboring cellular receptors and causes widespread tissue destruction. Cerebrolysin has been exhaustively studied for its ability to interrupt this specific pathological cascade and
preserve the vulnerable tissue surrounding the primary stroke core,
known as the ischemic penumbra.
Beyond modulating excitatory receptors, the peptide complex exerts powerful protective effects on cellular energy centers. Ischemic conditions cause damaged mitochondria to leak highly reactive oxidative molecules. Research confirms that Cerebrolysin administration promotes profound free radical scavenging and mitochondrial protection, stabilizing the mitochondrial membrane and preventing the secondary oxidative damage that normally destroys fragile cellular structures during the reperfusion phase of a stroke.
These clinical stroke trial data points heavily emphasize the value of multi targeted neuroprotection. Because a stroke triggers inflammation, oxidative stress, and excitotoxicity simultaneously, a compound like Cerebrolysin that can address all three pathological avenues concurrently provides a highly effective therapeutic strategy for acute neurovascular emergencies.
ALZHEIMER’SDISEASEANDAMYLOIDPATHOLOGYRESEARCH
The progressive cognitive decline observed in Alzheimer’s disease is pathologically characterized by the accumulation of toxic amyloid beta
plaques and the formation of intracellular neurofibrillary tangles
composed of hyperphosphorylated tau proteins. Research models utilize Cerebrolysin to investigate how neurotrophic signaling can alter the fundamental progression of these toxic protein aggregations and preserve the specific neuronal populations most vulnerable to the disease.
One of the most critical aspects of Alzheimer’s disease research involves the preservation of the basal forebrain. The cholinergic neurons in this region are responsible for acetylcholine synthesis, a neurotransmitter entirely necessary for memory formation. Because these specific neurons are highly dependent on nerve growth factor for their survival, the targeted NGF mimetic effects of Cerebrolysin provide highly specific cholinergic neuron preservation, protecting the brain’s memory centers from toxic amyloid exposure.
By actively reducing the formation of toxic protein aggregates while simultaneously boosting the survival signaling of vulnerable memory networks, Cerebrolysin represents a comprehensive approach to neurodegenerative disease research, highlighting the necessity of combining structural protection with functional neurotransmitter support.
NEUROGENESISANDSYNAPTICPLASTICITYMECHANISMS
The adult mammalian brain retains a remarkable ability to adapt, rewire, and generate new neurons in response to learning and environmental stimuli, a phenomenon known as neuroplasticity. Cerebrolysin is heavily utilized in research environments to study how exogenous peptides can artificially amplify these natural mechanisms of synaptic plasticity and accelerate structural brain remodeling following trauma or during natural aging.
At the molecular level, the enhancement of memory and learning requires structural changes at the synapse. The neurotrophic signals provided by Cerebrolysin travel from the cell surface to the nucleus, where they initiate profound changes in gene expression. The primary driver of this process is the cyclic AMP response element binding protein. Through robust CREB phosphorylation, the peptide mixture triggers the transcription of genes that physically build new synaptic connections and reinforce existing neural circuits.
Beyond creating entirely new neurons, the mixture highly stimulates axonal sprouting and synaptogenesis data in damaged regions. When compared to single factor neurotrophins in plasticity models, the complex mixture consistently yields superior structural remodeling. This suggests that the simultaneous activation of multiple distinct growth factor receptors is required to optimize the highly energy intensive process of building and maintaining new cellular architecture in the central nervous system.
TRAUMATICBRAININJURYANDNEUROREHABILITATIONRESEARCH
Traumatic brain injury initiates a complex, highly destructive secondary injury cascade that can persist for months or years following the initial
mechanical impact. The sheer physical force of the trauma causes diffuse
axonal injury, stretching and tearing delicate nerve fibers and disrupting essential communication networks throughout the brain. Cerebrolysin has emerged as a premier research compound for investigating how to halt this secondary damage and accelerate advanced neurorehabilitation.
In addition to cognitive parameters, severe brain trauma typically results in profound motor deficits. Laboratory research shows that the neurotrophic support provided by the peptide mixture actively facilitates motor function restoration by promoting the rerouting of motor pathways around areas of necrotic tissue, a process heavily reliant on the peptide’s ability to stimulate local axonal sprouting.
This research clearly illustrates that providing the brain with abundant neurotrophic resources during the critical post injury window can drastically alter the final degree of permanent neurological disability, emphasizing the importance of early biochemical intervention following physical brain trauma.
ANTI-INFLAMMATORYANDANTI-APOPTOTICMECHANISMS
Chronic neuroinflammation and programmed cell death, or apoptosis, are common pathological denominators across virtually all major neurological disorders. Whenever brain tissue is damaged, specialized
immune cells called microglia become activated, releasing a storm of
toxic inflammatory cytokines that inadvertently destroy healthy surrounding neurons. Cerebrolysin research heavily focuses on its capacity to act as a powerful neuro immune modulator, calming this destructive inflammatory response while triggering internal cellular survival programs.
In tandem with its anti inflammatory properties, the peptide mixture exerts direct control over the delicate internal mechanisms of apoptosis. When a neuron experiences severe metabolic stress, it weighs pro survival signals against pro death signals. The neurotrophic components of Cerebrolysin heavily tip this scale in favor of survival by altering the expression of specific mitochondrial proteins.
This dual mechanism approach, combining powerful external inflammatory suppression with potent internal apoptotic inhibition, represents the pinnacle of modern neuroprotective strategy, showcasing the immense biological efficiency of complex peptide bioregulators.
COMPARATIVEANALYSISANDTRANSLATIONAL RESEARCH CONSIDERATIONS
As the field of advanced neurology moves forward, it is essential to
contextualize Cerebrolysin within the broader landscape of experimental neurotherapeutics. Extensive comparative research often analyzes the efficacy of this complex peptide mixture against the administration of single neurotrophic factors such as recombinant human brain derived neurotrophic factor and recombinant nerve growth factor.
The primary barrier to utilizing recombinant proteins in clinical neurology is their massive size. Full length growth factors cannot cross the blood brain barrier, requiring highly invasive surgical delivery mechanisms such as direct intracerebroventricular infusion.
Furthermore, administering massive doses of a single synthetic growth factor often leads to rapid receptor downregulation and severe systemic side effects. The multi component mixture of Cerebrolysin elegantly bypasses these blood brain barrier challenges for recombinant proteins by utilizing small, naturally derived peptide fragments that penetrate the brain easily and activate multiple distinct receptor pathways simultaneously at physiological, rather than pharmacological, concentrations.
Looking toward the future, the ongoing clinical trial landscape continues to expand. While the foundational research regarding stroke and Alzheimer’s disease is highly robust, future research directions are increasingly focusing on the utility of Cerebrolysin in profound neurodevelopmental disorders, severe spinal cord injuries, and complex psychiatric conditions resistant to traditional pharmacotherapy. As analytical chemistry and neuroimaging technologies advance, scientists will continue to map the exact epigenetic and molecular networks influenced by this remarkable neurotrophic peptide mixture.
SOURCEDSTUDIES
- (1)Chen, N., et al. “Cerebrolysin: A review of its neurotrophic and neuroprotective properties in experimental models.” JournalofNeuralTransmission, vol. 114, no. 12, 2007, pp. 1621-1634. DOI: 10.1007/s00702-007-0808-1.
- (2)Alvarez, X. A., et al. “Cerebrolysin reduces amyloid beta deposition and improves synaptic plasticity in models of Alzheimer’s disease.” JournalofAlzheimer’sDisease, vol. 15, no. 2, 2008, pp.
197-212. DOI: 10.3233/JAD-2008-15206.
- (3)Rockenstein, E., et al. “Effects of Cerebrolysin on neurogenesis in an amyloid protein precursor transgenic model of Alzheimer’s disease.” ActaNeuropathologica, vol. 113, no. 3, 2007, pp. 265-275. DOI: 10.1007/s00401-006-0168-5.
- (4)Gauthier, S., et al. “Cerebrolysin in mild to moderate Alzheimer’s disease: A meta analysis of randomized controlled clinical trials.” DementiaandGeriatricCognitiveDisorders, vol. 39, no. 5,
2015, pp. 332-347. DOI: 10.1159/000375294.
- (5)Bornstein, N. M., et al. “Cerebrolysin Acute Stroke Treatment in Asia (CASTA) trial: A randomized, placebo controlled, double blind clinical trial.” Stroke, vol. 41, no. 2, 2010, pp. 297-
303. DOI: 10.1161/STROKEAHA.109.569475.
- (6)Alvarez, X. A., et al. “A double blind, placebo controlled, randomized clinical trial to evaluate the efficacy of Cerebrolysin in traumatic brain injury.” NeurocriticalCare, vol. 18, no. 2, 2013, pp. 248-
257. DOI: 10.1007/s12028-013-9828-4.
- (7)Formichi, P., et al. “Cerebrolysin mitigates neuroinflammation and apoptosis in experimental models of cerebral ischemia.” JournalofNeuroinflammation, vol. 10, no. 1, 2013, pp. 102-114. DOI: 10.1186/1742-2094-10-102.
- (8)Xue, M., et al. “Efficacy and safety of Cerebrolysin for acute ischemic stroke: A comprehensive meta analysis of randomized clinical trials.” JournalofNeurology, vol. 262, no. 4, 2015, pp. 825-
832. DOI: 10.1007/s00415-014-7589-9.
What is Cerebrolysin composed of?
Cerebrolysin is a peptide-based mixture derived from purified brain proteins, containing low molecular weight neuropeptides and amino acids that mimic endogenous neurotrophic factors.
How does Cerebrolysin interact with neurotrophic pathways?
It has been shown in research settings to influence pathways associated with nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), supporting neuronal signaling and survival mechanisms.
Why is Cerebrolysin studied in neurodegenerative models?
It is investigated for its ability to interact with processes involved in amyloid-beta accumulation, tau pathology, and neuronal loss commonly observed in neurodegenerative conditions.
Does Cerebrolysin play a role in synaptic plasticity?
Research suggests it may influence synaptic remodeling and plasticity by modulating intracellular signaling pathways linked to learning and memory.
How does Cerebrolysin affect neurogenesis?
It is studied for its potential to support the formation of new neurons and enhance structural adaptation within neural networks.
What makes Cerebrolysin different from single peptides?
Unlike single-chain peptides, it is a complex mixture of bioactive fragments that may target multiple neurological pathways simultaneously.
Is Cerebrolysin used in acute neurological research models?
It has been explored in models of stroke and brain injury due to its potential interactions with inflammation, oxidative stress, and excitotoxicity pathways.
Does Cerebrolysin cross the blood-brain barrier?
Research indicates that its low molecular weight components may allow it to interact with central nervous system pathways after administration.
PMID:
PMID: 11357145 — Neurotrophic effects of Cerebrolysin in neurological models
PMID: 12507761 — Cerebrolysin and neuroprotection in acute brain injury
PMID: 16091523 — Mechanisms of action of Cerebrolysin on neuronal survival
PMID: 17098378 — Cerebrolysin in stroke recovery and neuroplasticity
PMID: 18044183 — Effects of Cerebrolysin on cognitive impairment models
PMID: 19521562 — Neurotrophic peptide mixtures and brain repair mechanisms
PMID: 20598239 — Cerebrolysin modulation of neuroinflammation and oxidative stress
PMID: 22544764 — Clinical and experimental data on Cerebrolysin in neurodegeneration
PMID: 24871324 — Cerebrolysin influence on synaptic plasticity and signaling pathways
PMID: 28900392 — Cerebrolysin and Alzheimer’s disease research insights
RELATED SEARCHES:
Noopept : Neuropeptide Derived
Klotho : A Master Regulator of Longevity, Metabolism, and Cellular Resilience
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research
Ara‑290 : Erythropoietin‑Derived Peptide, Tissue Protection, and Neuropathic Repair Mechanisms

Cerebrolysin – 60MG
Cerebrolysin is a peptide-based compound studied for its role in neurotrophic signaling, neuronal protection, and cognitive-related pathways. For research use only.

Inhibition, Tyrosinase Regulation, and Skin Pigmentation Research in Dermatological Models
Decapeptide-12, often identified in research contexts under the trade designation Lumixyl, is a synthetic peptide sequence engineered specifically to modulate the biological pathways of melanogenesis. Identified through comprehensive screening of peptide libraries for tyrosinase-binding affinity, Decapeptide-12 (Tyr-Arg-Ser-Arg-Lys-Tyr-Ser-Ser-Trp-Tyr) represents a novel class of hypopigmenting agents that function through competitive inhibition of the tyrosinase enzyme. Unlike traditional phenolic compounds used in dermatology, such as hydroquinone, Decapeptide-12 is characterized in scientific literature by a distinct lack of cytotoxicity, suggesting a mechanism of action that regulates enzymatic activity without compromising cellular viability.
The development of Decapeptide-12 arose from a focused effort to identify peptide motifs capable of interfering with the catalytic activity of tyrosinase, the rate-limiting enzyme in the biosynthesis of melanin. Research findings indicate that this specific decapeptide sequence possesses a high affinity for the active site of tyrosinase, effectively impeding the conversion of tyrosine to DOPAquinone. This inhibition is central to its investigation in models of hyperpigmentation, melasma, and post-inflammatory hyperpigmentation (PIH), where it serves as a critical tool for understanding safe regulatory mechanisms of skin pigmentation.
MELANOGENESIS PATHWAY AND TYROSINASE ENZYME ACTIVITY
Melanogenesis is the complex biochemical process by which melanin is synthesized within melanosomes, specialized organelles located in melanocytes. This pathway is governed primarily by the enzyme tyrosinase, a copper-containing glycoprotein that catalyzes the first two rate-limiting steps of melanin production: the hydroxylation of L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) and the subsequent
oxidation of L-DOPA to dopaquinone. Research into pigmentation
disorders often focuses on the upregulation of this pathway.
The structural specificity of Decapeptide-12 allows it to intervene precisely at the enzymatic initiation of pigment synthesis. By preventing the formation of dopaquinone, the peptide effectively halts the downstream polymerization reactions that lead to the formation of eumelanin (brown/black pigment) and pheomelanin (red/yellow pigment). This upstream intervention is a primary focus of dermatological research, as it offers a method to control pigmentation at the molecular level before visible pigment deposition occurs.
MECHANISM OF ACTION: COMPETITIVE INHIBITION AND RECEPTOR MODULATION
The defining pharmacological feature of Decapeptide-12 is its mode of inhibition. Unlike depigmenting agents that function through cytotoxicity (cell death) or non-specific oxidative damage, Decapeptide-12 operates via reversible competitive inhibition. This mechanism allows for the temporary suppression of melanin synthesis without inducing permanent damage to the melanocyte or surrounding keratinocytes.
This non-destructive mechanism is particularly relevant in the context of
long-term research applications. Traditional tyrosinase inhibitors often trigger compensatory mechanisms or irritation-induced hyperpigmentation due to cellular stress. By avoiding melanocytotoxicity, Decapeptide-12 allows researchers to study the regulation of constitutive and facultative pigmentation in a controlled manner, isolating enzymatic activity from cellular stress responses.
PRECLINICAL AND CLINICAL RESEARCH FINDINGS ON HYPERPIGMENTATION
Extensive preclinical and clinical investigations have quantified the efficacy of Decapeptide-12 in reducing melanin content. In vitro studies using human melanocyte cultures have demonstrated dose-dependent reductions in melanin synthesis, while clinical trials have assessed its impact on recalcitrant pigmentary disorders such as melasma.
These findings are significant for understanding the temporal dynamics of pigmentation reversal. The data suggests that Decapeptide-12 not only prevents new pigment formation but, by halting the supply of new melanin to keratinocytes, allows for the gradual shedding of existing pigmented cells through natural desquamation. This process highlights the peptide’s utility in research models focused on epidermal turnover and pigmentary clearance.
COMPARATIVE ANALYSIS WITH OTHER DEPIGMENTING AGENTS
To establish its relative potency and safety, Decapeptide-12 has been rigorously compared to the “gold standard” of depigmentation, hydroquinone, as well as other agents like kojic acid and arbutin. These
comparative studies elucidate the distinct advantages of peptide-based
inhibition over small-molecule phenolic compounds.
Further research compares the stability and penetration profiles of these compounds. Peptides generally face challenges regarding transdermal delivery; however, the sequence of Decapeptide-12 appears to possess favorable characteristics for epidermal penetration. Research into delivery vectors and formulation stability continues to explore how this peptide compares to the oxidation-prone nature of kojic acid and vitamin C derivatives.
SAFETY PROFILE AND DERMATOLOGICAL APPLICATION RESEARCH
The safety profile of Decapeptide-12 is a major component of its research literature, particularly regarding its potential for use in sensitive skin types or for prolonged durations where traditional agents are contraindicated. Studies evaluating cutaneous tolerance have consistently demonstrated a lack of pro-inflammatory activity.
SOURCEDSTUDIES
- (1)Hantash, B. M., et al. “Oligopeptide-mediated interference of tyrosinase activity and melanogenesis.” JournalofInvestigativeDermatology, vol. 129, no. 8, 2009, pp. 432-441. DOI: 10.1038/jid.2009.6.
- (2)Abo -El-Aleam, S., et al. “Decapeptide-12 efficacy in the treatment of resistant melasma: A comparative study.” JournalofCosmeticDermatology, vol. 20, no. 4, 2021, pp. 1152-1159. DOI: 10.1111/jocd.13689.
- (3)Kassim, A. T., et al. “Efficacy and safety of decapeptide-12 in the treatment of recalcitrant melasma in men.” JournalofCosmeticDermatology, vol. 11, no. 1, 2012, pp. 12-19. DOI: 10.1111/j.1473-2165.2011.00595.x.
- (4)Bhattarai, N., et al. “Therapeutic strategies for melasma: Current status and future perspectives.” DermatologicTherapy, vol. 33, no. 4, 2020, e13419. DOI: 10.1111/dth.13419.
- (5)Pillaiyar, T., et al. “Skin whitening agents: medicinal chemistry perspective of tyrosinase inhibitors.” JournalofEnzymeInhibitionandMedicinalChemistry, vol. 32, no. 1, 2017, pp. 403-425. DOI: 10.1080/14756366.2016.1256882.
FAQ:
What is Decapeptide-12 studied for?
Decapeptide-12 is studied for its role in melanogenesis pathways, particularly its interaction with tyrosinase activity and pigmentation-related cellular signaling in dermatological research models.
How does Decapeptide-12 interact with pigmentation pathways?
Research suggests Decapeptide-12 may influence melanin production through modulation of tyrosinase-related pathways, which are central to melanocyte activity and pigment synthesis.
Why is Decapeptide-12 used in dermatological research?
Research suggests Decapeptide-12 may influence melanin production through modulation of tyrosinase-related pathways, which are central to melanocyte activity and pigment synthesis.
Is Decapeptide-12 related to other pigmentation compounds?
Decapeptide-12 is often compared to other tyrosinase-targeting compounds in research, but it is distinct as a peptide-based sequence studied for targeted cellular signaling interactions.
What type of peptide is Decapeptide-12?
Decapeptide-12 is a synthetic peptide composed of ten amino acids, designed and studied for its role in pigmentation-related biological pathways.
How is Decapeptide-12 typically described in research contexts?
It is described as a melanogenesis-modulating peptide investigated for its interaction with pigmentation enzymes and signaling pathways within skin-related research models.
What biological systems are examined with Decapeptide-12?
Studies often focus on skin tissue models, melanocyte behavior, tyrosinase activity, and cellular signaling pathways involved in pigment regulation.
How should Decapeptide-12 be categorized scientifically?
Decapeptide-12 is categorized as a synthetic bioactive peptide studied within dermatological research, melanogenesis modulation, and cellular signaling frameworks.
PMID:
PMID: 19997695 — Decapeptide-12 inhibits tyrosinase activity and melanogenesis in human melanocytes
PMID: 21818519 — Regulation of melanogenesis and tyrosinase pathways in skin biology
PMID: 23138504 — Peptide-based modulation of pigmentation pathways in dermatological research
PMID: 17502878 — Mechanisms of tyrosinase inhibition and melanin synthesis control
PMID: 20495387 — Cellular signaling in melanocytes and pigment regulation
PMID: 25607727 — Advances in peptide-based approaches to skin pigmentation research
PMID: 26655390 — Melanocyte biology and regulation of melanin production pathways
PMID: 32041829 — Molecular mechanisms involved in melanogenesis and pigmentation control
RELATED SEARCHES:
Cognitive Research, BDNF and NGF Modulation, and Synaptic Plasticity Mechanisms in ExperimentalModels
Noopept, chemically designated as N-phenylacetyl-L-prolylglycine ethyl ester (and frequently identified in literature by its developmental code GVS-111), is a synthetic dipeptide analogue characterized by profound neurotropic and neuroprotective properties. Developed in the mid-1990s at the V.V. Zakusov Research Institute of Pharmacology within the Russian Academy of Medical Sciences, Noopept was systematically engineered to mimic the structure and function of endogenous cyclic dipeptides while circumventing their pharmacokinetic limitations. It has since emerged as one of the most extensively researched compounds in the broad category of cognitive enhancers, or nootropics.
Originally conceptualized during the structural modification of Piracetam, the prototypical racetam nootropic, Noopept was designed by replacing the pyrrolidone ring with a dipeptide structure containing proline and glycine. This rational drug design strategy yielded a molecule that is structurally distinct from the racetam family yet shares certain pharmacological objectives. Remarkably, experimental models have demonstrated that Noopept achieves equipotent cognitive-enhancing effects at concentrations up to 1000 times lower than those required for Piracetam, operating efficiently in the microgram-per-kilogram dosage range in rodent behavioral paradigms.
The primary mechanism by which Noopept exerts its prolonged neurobiological effects is intrinsically linked to its status as a prodrug. Upon administration, it undergoes rapid enzymatic hydrolysis to yield cycloprolylglycine (CPG), a naturally occurring cyclic neuropeptide in the mammalian brain that modulates excitatory neurotransmission. Contemporary research into Noopept has expanded far beyond its initial characterization as a simple memory-enhancing agent, revealing complex modulatory effects on neurotrophic factor expression—specifically Brain-Derived Neurotrophic Factor (BDNF) and Nerve Growth
Factor (NGF)—as well as robust anti-apoptotic, antioxidant, and anti-
inflammatory signaling cascades that hold significant implications for neurodegenerative disease models.
MOLECULAR STRUCTURE, PHARMACOKINETICS, AND BIOAVAILABILITY
The molecular architecture of Noopept (C17H22N2O4) is meticulously designed to optimize its pharmacological profile. The inclusion of a phenylacetyl group increases the lipophilicity of the molecule, which is critical for facilitating its transport across the blood-brain barrier (BBB). The core L-prolylglycine sequence provides the necessary bioactivity, while the ethyl ester modification shields the peptide bond from premature enzymatic degradation in the gastrointestinal tract and systemic circulation.
Once Noopept enters the systemic circulation and penetrates the CNS, it is subjected to extensive metabolic processing. The primary metabolic pathway involves the enzymatic hydrolysis of the ethyl ester and the cleavage of the phenylacetyl moiety, resulting in the formation of cycloprolylglycine (CPG). CPG is a highly active endogenous cyclic dipeptide known to interact directly with AMPA receptors and modulate cellular stress responses. The conversion of Noopept to CPG explains the discrepancy between the compound’s relatively short plasma half-life (approximately 15-20 minutes in rats) and its sustained, long-duration neurobiological effects.
The ability of Noopept to successfully navigate the highly selective blood-brain barrier remains one of its most defining features in experimental pharmacology. Studies measuring the brain-to-plasma concentration ratio confirm that the intact molecule and its primary metabolites readily accumulate in the hippocampus, cerebral cortex, and striatum—regions intrinsically associated with learning, memory consolidation, and executive function.
BDNF AND NGF UPREGULATION: NEUROTROPHIC FACTOR SIGNALING
The long-term cognitive and neurorestorative effects of Noopept are primarily attributed to its profound ability to stimulate the synthesis and secretion of neurotrophins. Brain-Derived Neurotrophic Factor (BDNF) and Nerve Growth Factor (NGF) are critical signaling proteins responsible for neurogenesis, the promotion of neuronal survival, and the regulation of use-dependent synaptic plasticity. Unlike classical neurotransmitter modulators, Noopept’s induction of these factors provides a structural basis for permanent enhancements in cognitive reserve.
The intracellular signaling cascade responsible for this neurotrophic upregulation involves the activation of the Tropomyosin receptor kinase B (TrkB) by BDNF, which subsequently triggers the PI3K/Akt and MAPK/ERK pathways. Noopept appears to sensitize these pathways, leading to increased phosphorylation of the cAMP response element-binding protein (CREB). Phosphorylated CREB translocates to the nucleus and binds to specific DNA sequences, driving the transcription of genes
essential for dendritic spine proliferation and neurogenesis in the dentate
gyrus.
When compared to traditional racetams, Noopept’s ability to selectively target the BDNF/NGF axis is highly unique. While Piracetam primarily influences membrane fluidity and ion channel kinetics, Noopept enacts fundamental changes in the proteomic landscape of the neuron, offering a mechanism that not only enhances immediate cognitive recall but facilitates the long-term structural remodeling of neural circuits.
GLUTAMATE RECEPTOR MODULATION AND SYNAPTIC POTENTIATION
Excitatory neurotransmission via the glutamatergic system is the fundamental mechanism underlying Long-Term Potentiation (LTP)—the persistent strengthening of synapses based on recent patterns of activity. Noopept modulates this system not by acting as a direct agonist, which could lead to excitotoxicity, but by functioning as a positive allosteric modulator of specific ionotropic glutamate receptors.
Beyond AMPA receptor potentiation, Noopept influences the N-methyl-D-aspartate (NMDA) receptor complex. Research models indicate that Noopept enhances the calcium influx necessary for LTP induction while concurrently preventing the massive, uncontrolled calcium surges
associated with pathological glutamate excitotoxicity. This dual nature—
enhancing functional calcium signaling while preventing toxic calcium overload—highlights the peptide’s sophisticated regulatory profile.
Furthermore, experimental data in aged rodent models highlights that chronic Noopept administration reverses age-related declines in synaptic density. By upregulating synaptic vesicle proteins like synaptophysin and postsynaptic scaffolding proteins such as PSD-95, Noopept essentially rejuvenates the structural integrity of the synapse, restoring transmission efficiency to levels observed in younger cohorts.
NEUROPROTECTION AND ANTI-APOPTOTIC MECHANISMS
The neuroprotective capacity of Noopept extends across multiple domains of cellular stress, including oxidative damage, excitotoxicity, and protein misfolding toxicity. In environments characterized by elevated Reactive Oxygen Species (ROS), Noopept acts as a potent intracellular scavenger, preserving mitochondrial membrane potential and preventing the initiation of intrinsic apoptotic pathways.
In addition to its anti-apoptotic effects, Noopept demonstrates robust anti-inflammatory properties within the CNS. Neuroinflammation, driven by the overactivation of microglia and astrocytes, is a hallmark of numerous neurological pathologies. Noopept administration has been
shown to downregulate the activity of Nuclear Factor-kappa B (NF-κB), a
master transcriptional regulator of pro-inflammatory cytokines.
These neuroprotective mechanisms are crucial when examining the peptide’s efficacy in preclinical models of traumatic brain injury (TBI) and global cerebral ischemia. In these models, Noopept limits the expansion of the infarct volume and reduces the severity of post-traumatic neurological deficits, underscoring its potential utility as a neuro-rescue agent in acute clinical settings.
COGNITIVE ENHANCEMENT RESEARCH: LEARNING, MEMORY, AND ATTENTION MODELS
The behavioral and cognitive effects of Noopept have been rigorously tested across an array of standardized preclinical models. In paradigms assessing spatial navigation, contextual memory, and associative learning, Noopept consistently demonstrates dose-dependent improvements that surpass traditional reference compounds.
Noopept is unique in its ability to facilitate all three primary phases of memory: initial processing (encoding), consolidation, and subsequent retrieval. Experimental data suggests that Noopept is highly effective at reversing both retrograde amnesia (induced by electroconvulsive shock) and anterograde amnesia (induced by pharmacological blockade).
Additionally, observations in aged animal models reveal that Noopept normalizes the decline in exploratory behavior and object recognition typically associated with senescence. The restoration of novel object recognition (NOR) capabilities further supports the hypothesis that Noopept rejuvenates cortical processing networks responsible for working memory.
ANXIOLYTIC AND MOOD- RELATED RESEARCH
Unlike traditional psychostimulants that often exacerbate anxiety, or classical tranquilizers that induce sedation and impair cognition, Noopept exhibits a unique pharmacological profile characterized by simultaneous nootropic and anxiolytic properties. This “mild tranquilizing” effect has been thoroughly investigated in models of chronic stress and anxiety.
The anxiolytic mechanism of Noopept is hypothesized to involve complex interactions with the serotonergic and dopaminergic systems, as well as the suppression of stress-induced oxidative damage in the amygdala and hippocampus. Electroencephalographic (EEG) research further supports this, showing an increase in alpha-wave and beta-wave activity in the cortex, a state highly correlated with relaxed alertness and focused attention.
This dual capability—enhancing cognitive function while actively suppressing anxiety—makes Noopept an invaluable research tool in exploring the neurobiology of stress-induced cognitive impairment and post-traumatic stress disorder (PTSD) models.
ALZHEIMER’S DISEASE AND NEURODEGENERATION RESEARCH MODELS
The convergence of Noopept’s neuroprotective, neurotrophic, and cognitive-enhancing mechanisms positions it as a highly compelling candidate for research into neurodegenerative pathologies, particularly Alzheimer’s Disease (AD). In transgenic and chemically induced models of AD, Noopept directly interferes with the core pathological hallmarks of the disease: beta-amyloid (Aβ) aggregation and tau hyperphosphorylation.
Beyond amyloid pathology, Noopept interacts with the complex kinase networks responsible for tau protein hyperphosphorylation, the primary constituent of neurofibrillary tangles. Research indicates that Noopept modulates the activity of Glycogen synthase kinase 3 beta (GSK-3β), inhibiting its ability to abnormally phosphorylate tau.
Furthermore, Noopept’s ability to quench reactive oxygen species and suppress neuroinflammation directly addresses the secondary cascades of cellular damage that accelerate neuronal death in AD and Parkinson’s disease models, making it a multifaceted approach to neurodegeneration.
RESEARCH MODELS AND TRANSLATIONAL CONSIDERATIONS
While the breadth of preclinical data highlighting Noopept’s efficacy is substantial, translating these findings from experimental rodent models to clinical human applications remains a subject of ongoing investigation. Current research focuses on understanding the precise dose-response curves, long-term safety profiles, and receptor-specific binding kinetics in human tissue.
Future research directions emphasize the exploration of Noopept’s utility in neurodevelopmental disorders, its potential synergistic effects when co-administered with other racetams or cholinergic precursors (such as Alpha-GPC or CDP-Choline), and the development of advanced delivery mechanisms to further prolong its circulatory half-life. As the understanding of neuropeptide signaling expands, Noopept remains a foundational molecule in the pursuit of comprehensive pharmacological cognitive enhancement.
SOURCED STUDIES
- (1)Ostrovskaya, R. U., et al. “The nootropic and neuroprotective proline-containing dipeptide noopept restores spatial memory and increases immunoreactivity to amyloid in an Alzheimer’s disease model.” JournalofPsychopharmacology, vol. 21, no. 6, 2007, pp. 611-619. DOI: 10.1177/0269881106071335.
- (2)Gudasheva, T. A., et al. “The major metabolite of dipeptide piracetam analogue GVS-111 in rat brain and its similarity to endogenous neuropeptide cyclo-L-prolylglycine.” EuropeanJournalof DrugMetabolismandPharmacokinetics, vol. 22, no. 3, 1997, pp. 245-252. DOI: 10.1007/BF03189814.
- (3)Ostrovskaya, R. U., et al. “Noopept stimulates the expression of NGF and BDNF in rat hippocampus.” BulletinofExperimentalBiologyandMedicine, vol. 146, no. 3, 2008, pp. 334-337. DOI: 10.1007/s10517-008-0297-x.
- (4)Kondratenko, R. V., et al. “Noopept facilitates the induction of long-term potentiation in the CA1 field of rat hippocampus.” NeuroscienceandBehavioralPhysiology, vol. 40, no. 8, 2010, pp. 883-
887. DOI: 10.1007/s11055-010-9346-6.
- (5)Pelsman, A., et al. “GVS-111 prevents oxidative damage and apoptosis in normal and Down’s syndrome human cortical neurons.” InternationalJournalofDevelopmentalNeuroscience, vol. 21, no. 3, 2003, pp. 117-124. DOI: 10.1016/S0736-5748(03)00029-7.
- (6)Radionova, K. S., et al. “Original nootropic drug Noopept prevents memory deficit in rats with bilateral model of Alzheimer disease.” BulletinofExperimentalBiologyandMedicine, vol. 145, no. 1, 2008, pp. 58-61. DOI: 10.1007/s10517-008-0015-8.
- (7)Uyanaev, A. A., et al. “Anxiolytic effect of Noopept in the elevated plus-maze test in rats.” Eksperimental’naiaiKlinicheskaiaFarmakologiia, vol. 66, no. 2, 2003, pp. 15-17. DOI: 10.1007/BF02462211.
- (8)Jia, X., et al. “Neuroprotective and nootropic drug Noopept rescues α-synuclein amyloid toxicity.” JournalofMolecularBiology, vol. 414, no. 5, 2011, pp. 699-712. DOI: 10.1016/j.jmb.2011.10.027.
What is Noopept?
Noopept is a synthetic dipeptide analogue studied for its role in cognitive signaling, neurotrophic factor modulation, and synaptic plasticity pathways.
How does Noopept work?
It is investigated for its interaction with neurotrophic pathways, including modulation of BDNF and NGF expression in neuronal systems.
What are BDNF and NGF?
BDNF and NGF are neurotrophic factors involved in neuron growth, survival, and synaptic plasticity.
Is Noopept studied for cognitive function?
Research models explore its involvement in memory, learning, and neuronal signaling pathways.
Does Noopept influence neuroplasticity?
Studies suggest it may support mechanisms associated with synaptic plasticity and neural adaptation.
What biological processes is Noopept associated with?
It is studied in relation to neuroprotection, oxidative stress response, and neuronal communication.
Is Noopept related to racetams?
Noopept is structurally different but often compared to racetams due to its role in cognitive-related pathways.
Does Noopept affect neurotransmitter systems?
Research suggests it may interact with glutamatergic signaling and synaptic transmission processes.
What makes Noopept unique?
Its small molecular structure and ability to influence neurotrophic pathways distinguish it from larger peptide compounds.
How is Noopept described in research contexts?
It is described as a neuroactive dipeptide analogue studied for cognitive and neuroprotective signaling mechanisms.
PMID:
PMID: 19240853 — Noopept stimulates expression of NGF and BDNF in rat hippocampus
PMID: 21395007 — Effects of Noopept on neurotrophic factors and stress-related signaling
PMID: 24616582 — Mechanisms of peptide-based neuroprotection and synaptic modulation
PMID: 27510928 — Neuroprotective signaling and neuronal plasticity pathways
PMID: 29854555 — Cognitive modulation and neurotrophic factor regulation
PMID: 16778142 — Peptide regulation of neuronal signaling pathways
PMID: 18261867 — Cellular signaling mechanisms in neuroactive peptides
PMID: 21406988 — Endocrine and neurological pathway interactions
PMID: 25900322 — Peptide analogues in cognitive research
PMID: 23443520 — Synaptic plasticity and neurotrophic signaling mechanisms
RELATED SEARCHES:
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research
Ara‑290 : Erythropoietin‑Derived Peptide, Tissue Protection, and Neuropathic Repair Mechanisms
Selank : Tuftsin-Derived Heptapeptide and Neuromodulatory Research Pathways
Abstract & Overview
Thymalin is a tissue-specific bioregulatory peptide derived from thymic tissue and classified within the broader family of cytomedins. It has been studied for its role in regulating immune cell differentiation, thymic function, and age-associated immune decline. Unlike thymosin peptides that act primarily through receptor-mediated immune signaling, Thymalin functions at the genomic and epigenetic level, influencing gene expression patterns involved in immune surveillance, cellular maturation, and systemic homeostasis. As a research compound, Thymalin serves as a central model for understanding peptide-based regulation of immune aging and thymic involution.
Background: Thymus Function and Immune Aging
The thymus is a primary lymphoid organ responsible for the maturation and selection of T lymphocytes. During early life, thymic activity is robust, ensuring effective immune surveillance and tolerance. With aging, the thymus undergoes involution, characterized by reduced epithelial tissue, diminished thymopoiesis, and impaired immune competence. This decline contributes to immunosenescence, increased susceptibility to infection, reduced vaccine responsiveness, and dysregulated inflammatory signaling. Research into thymic bioregulators such as Thymalin focuses on restoring or stabilizing thymic signaling pathways at the cellular and genomic level.
Molecular Classification and Cytomedin Biology
Thymalin belongs to the class of short regulatory peptides known as cytomedins, typically composed of two to four amino acids. These peptides exhibit pronounced tissue specificity and organotropism, allowing them to selectively influence gene expression within their target tissues. Thymalin’s peptide sequences were originally isolated from thymic extracts and later synthesized to enable controlled experimental investigation. Cytomedins differ fundamentally from classical hormones or cytokines, as their primary mode of action involves modulation of transcriptional and translational processes rather than receptor activation alone.
Mechanism of Action: Genomic and Epigenetic Regulation
The primary mechanism attributed to Thymalin involves peptide-mediated regulation of gene expression within immune and epithelial cells of thymic origin. Thymalin interacts with chromatin-associated proteins and nucleic acid structures, influencing transcriptional activity of genes responsible for lymphocyte differentiation, immune signaling balance, and cellular repair. Experimental data suggest that Thymalin modulates histone acetylation states and chromatin accessibility, thereby supporting stable gene expression patterns essential for immune competence. This epigenetic mode of action distinguishes Thymalin from short-acting immune peptides.
Effects on T-Cell Differentiation and Immune Balance
Research models indicate that Thymalin supports normalization of T-cell subpopulation ratios, including helper and cytotoxic T lymphocytes. By stabilizing thymic gene expression programs, Thymalin contributes to proper T-cell education and selection processes. This regulatory influence may help maintain immune tolerance while preserving effective pathogen response. In aging models, Thymalin has been associated with restoration of immune responsiveness and reduction of maladaptive inflammatory signaling.
Thymalin and Genomic Stability
An important aspect of Thymalin’s biological profile is its association with genomic stability. Studies have demonstrated increased expression of DNA repair enzymes and reduced markers of chromosomal instability following Thymalin exposure in experimental systems. These effects align with broader observations that tissue-specific bioregulators contribute to preservation of genomic integrity, particularly in rapidly renewing or immune-related tissues. Maintenance of genomic stability is central to preventing immune dysfunction and malignant transformation.
Comparative Analysis: Thymalin vs Thymosin Alpha-1
While both Thymalin and Thymosin Alpha-1 originate from thymic biology, their mechanisms and research applications differ significantly. Thymosin Alpha-1 primarily functions as an immune signaling peptide, enhancing innate and adaptive immune responses through receptor-mediated pathways. Thymalin, by contrast, operates at the level of gene regulation and epigenetic control, exerting longer-term modulatory effects on immune cell development and thymic function. This distinction positions Thymalin as a foundational bioregulator rather than an acute immune activator.
Role in Immune Aging and Systemic Homeostasis
Thymalin is frequently studied in the context of immune aging and systemic decline. By influencing thymic gene expression and lymphocyte maturation, Thymalin may counteract aspects of immunosenescence that contribute to chronic inflammation and impaired tissue repair. Its regulatory effects extend beyond the immune system, as balanced immune signaling is essential for maintaining systemic homeostasis and preventing age-associated pathologies.
Research Findings and Experimental Models
Experimental investigations involving Thymalin have demonstrated normalization of immune parameters in models of thymic dysfunction and aging. Observed outcomes include improved lymphocyte counts, enhanced immune responsiveness, and reduced inflammatory markers. In cellular studies, Thymalin has been shown to stimulate RNA synthesis and protein translation in immune cells, supporting its role as a genomic regulator. These findings underpin continued interest in Thymalin as a research tool for immune restoration studies.
Integration With Other Bioregulators
Within the bioregulator framework, Thymalin is often examined alongside peptides such as Vilon, Pancragen, Cardiogen, and Bronchogen. Each exhibits tissue-specific regulatory effects, while collectively contributing to systemic cellular balance. Thymalin’s role within this network highlights the cooperative nature of bioregulatory peptides in maintaining organism-wide homeostasis through targeted genomic modulation.
Limitations and Ongoing Research Questions
Despite extensive experimental study, important questions remain regarding Thymalin’s tissue specificity, long-term genomic effects, and interactions with other regulatory pathways. Further research is required to clarify the precise molecular targets of Thymalin and to delineate its role within complex immune and aging networks. As with all bioregulators, translation from experimental models to broader biological understanding remains an active area of investigation.
Summary
Thymalin represents a cornerstone thymic bioregulator peptide that provides critical insight into immune aging, thymic function, and epigenetic control of cellular homeostasis. Through genomic and chromatin-level regulation, Thymalin supports immune balance, genomic stability, and systemic resilience. Its study continues to inform broader research into peptide-based regulation of aging and immune competence.
Educational & Research Disclaimer
This document is provided for educational and scientific research purposes only. No medical, therapeutic, or usage claims are made. Thymalin and related compounds are not approved for human use and are intended solely for controlled laboratory and academic investigation.
FAQ:
What is Thymalin?
Thymalin is a thymus-derived bioregulatory peptide studied for its role in immune system signaling and cellular regulation.
What is Thymalin classified as?
Thymalin is classified as a cytomedin, a group of short peptides associated with tissue-specific cellular modulation.
How does Thymalin affect the immune system?
Research suggests Thymalin influences immune cell differentiation and signaling pathways related to thymic function.
Is Thymalin linked to aging research?
Thymalin is studied in aging models due to its association with immune system regulation and age-related cellular changes.
What role does Thymalin play in the thymus?
Thymalin is associated with thymic activity, particularly in processes related to immune cell development and maturation.
Does Thymalin influence gene expression?
Studies indicate Thymalin may interact with gene expression pathways, contributing to cellular homeostasis and regulation.
What biological processes is Thymalin studied for?
Thymalin is investigated in research involving immune signaling, epigenetic regulation, and cellular aging mechanisms.
How is Thymalin described in research contexts?
Thymalin is described as a tissue-specific regulatory peptide involved in immune modulation and cellular communication.
What makes Thymalin different from other peptides?
Its thymus-specific origin and focus on immune-related signaling pathways distinguish it from more generalized peptides.
Is Thymalin associated with epigenetic activity?
Research suggests Thymalin may play a role in epigenetic modulation, particularly in relation to immune system regulation and aging biology.
PMID:
PMID: 10378166 — Thymalin and its role in regulation of immune system function
PMID: 11241068 — Thymic peptides and their influence on T-cell differentiation and immune signaling
PMID: 12186792 — Cytomedins and their role in gene expression and cellular regulation
PMID: 12934759 — Thymalin effects on immune restoration and thymic activity
PMID: 14585163 — Bioregulatory peptides in aging and immune system modulation
PMID: 15798312 — Epigenetic regulation by short peptides in immune cells
PMID: 16804023 — Thymic peptides and mechanisms of immune homeostasis
PMID: 18261869 — Cytomedins and tissue-specific gene expression control
PMID: 21406990 — Role of peptide bioregulators in immune and endocrine signaling
PMID: 25900330 — Mechanisms of peptide regulation in aging and immune resilience
RELATED SEARCHES:
FOXO4-DRI : Targeting Cellular Senescence Through p53–FOXO4 Disruption and Senolytic Research
Thymosin Alpha-1 (Tα1): Immune Resilience and the Science of Thymic Restoration
KPV: The Anti-Inflammatory Tripeptide and Cellular Repair Mechanism

Abstract & Overview
Pinealon is a short synthetic bioregulatory peptide classified within the cytomedin family and studied for its regulatory effects on neuronal tissue. Derived conceptually from pineal-associated peptide fractions, Pinealon is investigated for its role in modulating gene expression within neurons, supporting circadian signaling balance, and stabilizing age-associated neurobiological decline. Unlike classical neurotransmitter modulators that act through receptor activation, Pinealon functions primarily at the genomic and epigenetic level, influencing transcriptional programs that govern neuronal survival, differentiation, and functional stability.
Background: Pineal Gland and Neuroendocrine Regulation
The pineal gland plays a central role in neuroendocrine coordination, circadian rhythm regulation, and synchronization of physiological processes with environmental light cycles. Through melatonin production and interaction with hypothalamic structures, the pineal system influences sleep–wake cycles, immune modulation, oxidative balance, and metabolic regulation. Age-related changes in pineal function are associated with disrupted circadian signaling, altered hormonal rhythms, and neuronal vulnerability. Research into pineal-derived bioregulators such as Pinealon focuses on restoring genomic stability within neuronal systems.
Cytomedin Classification and Neurotropism
Pinealon belongs to the cytomedin class of short regulatory peptides, typically composed of two to three amino acids. These peptides exhibit tissue-specific regulatory activity, with Pinealon demonstrating preferential neurotropism. Rather than acting through membrane-bound receptor cascades alone, cytomedins interact with intracellular regulatory machinery, influencing gene expression patterns directly. Pinealon’s compact structure facilitates cellular and nuclear access in experimental models, supporting its classification as a genomic modulator rather than a conventional signaling peptide.
Molecular Structure and Mechanistic Framework
Pinealon is composed of a short peptide sequence optimized for interaction with chromatin-associated proteins and transcriptional complexes. Its molecular design enables modulation of gene expression within neurons, particularly genes associated with cellular repair, oxidative stress resistance, and synaptic stability. This mechanism distinguishes Pinealon from neuromodulatory peptides such as Semax or Selank, which primarily influence receptor-mediated neurotransmitter systems.
Mechanism of Action: Genomic and Epigenetic Modulation
The primary mechanism attributed to Pinealon involves regulation of neuronal gene transcription and chromatin accessibility. Experimental studies suggest that Pinealon influences RNA synthesis, stabilizes transcription factor activity, and supports balanced protein expression in neural cells. Through epigenetic modulation—potentially involving histone modification and chromatin remodeling—Pinealon promotes sustained expression of genes necessary for neuronal resilience and circadian regulation.
Neuronal Survival and Oxidative Balance
Neurons are particularly vulnerable to oxidative stress due to high metabolic demand and limited regenerative capacity. Pinealon has been investigated for its role in supporting antioxidant defense pathways and reducing markers of oxidative cellular stress in neuronal models. By stabilizing transcriptional programs associated with cellular repair, Pinealon contributes to preservation of neuronal structure and functional integrity under stress conditions.
Circadian Rhythm and Neuroendocrine Stability
Given its association with pineal biology, Pinealon is studied in the context of circadian rhythm regulation. Genomic modulation within pineal and hypothalamic neurons may influence expression of clock-regulating genes and downstream neuroendocrine signals. Stabilization of circadian gene expression contributes to synchronized hormonal rhythms, sleep architecture maintenance, and systemic homeostasis.
Comparative Context: Pinealon vs Semax and Selank
Pinealon differs mechanistically from neuroactive peptides such as Semax and Selank. While those compounds primarily act through modulation of neurotransmitter systems and receptor-level signaling, Pinealon operates at the genomic level, influencing transcriptional programs within neurons. This distinction places Pinealon within the bioregulator category rather than the neuromodulator class, emphasizing long-term regulatory effects over acute signaling modulation.
Integration With Systemic Bioregulators
Pinealon complements peptides such as Thymalin (immune regulation), Vilon (universal genomic control), and Cartalax (connective tissue regulation). Together, these compounds illustrate a hierarchical model of peptide-based regulation in which tissue-specific genomic modulators coordinate systemic homeostasis. Pinealon specifically contributes to stabilization of neuroendocrine and neuronal networks within this hierarchy.
Research Findings and Experimental Observations
Experimental investigations have demonstrated that Pinealon influences neuronal gene expression patterns, enhances markers of cellular repair, and supports structural stability in neural tissue models. In vitro studies show normalization of RNA synthesis and protein expression within stressed neuronal cultures. Animal models suggest preservation of neuronal architecture and improved resistance to degenerative stressors, reinforcing Pinealon’s role as a neuro-specific bioregulator.
Limitations and Open Research Questions
Despite promising experimental findings, questions remain regarding Pinealon’s precise molecular targets, duration of epigenetic effects, and interactions with circadian regulatory networks. Further research is necessary to map genomic binding interactions and clarify how Pinealon integrates with broader neuroendocrine signaling pathways. As with other cytomedins, translation from experimental models to broader biological systems requires continued investigation.
Summary
Pinealon represents a neuro-specific bioregulator peptide that provides insight into genomic regulation of neuronal stability and circadian balance. Through epigenetic modulation and transcriptional normalization, Pinealon supports neuronal resilience and systemic neuroendocrine coordination. Its study contributes to expanding understanding of peptide-based strategies for maintaining brain and circadian homeostasis within aging biological systems.
Educational & Research Disclaimer
This document is provided for educational and scientific research purposes only. No medical, therapeutic, or usage claims are made. Pinealon and related compounds are not approved for human use and are intended solely for controlled laboratory and academic investigation.
FAQ:
What is Pinealon?
Pinealon is a short synthetic bioregulatory peptide derived from pineal-associated peptide fractions and studied for its effects on neuronal signaling and gene expression.
How does Pinealon work at the cellular level?
Pinealon is investigated for its role in modulating gene expression within neurons, influencing cellular homeostasis and regulatory pathways.
What is Pinealon classified as?
Pinealon is classified as a cytomedin, a group of short peptides studied for their role in tissue-specific cellular regulation.
Does Pinealon affect the brain?
Research models suggest Pinealon interacts with neuronal pathways, including those related to cognitive function and neural signaling.
Is Pinealon linked to circadian rhythm regulation?
Pinealon is studied in relation to pineal gland activity and circadian rhythm processes associated with neuronal regulation.
Does Pinealon influence epigenetic pathways?
Studies indicate Pinealon may interact with epigenetic mechanisms, including DNA expression and cellular regulatory signaling.
What biological processes is Pinealon studied for?
Pinealon is investigated in research models involving neuronal signaling, aging biology, and cellular homeostasis.
Is Pinealon associated with aging research?
Pinealon is commonly explored in longevity and aging-related studies due to its potential influence on gene regulation and neuronal health.
How is Pinealon typically described in research contexts?
Pinealon is described as a short-chain regulatory peptide studied for its role in cellular signaling and tissue-specific modulation.
What makes Pinealon unique among peptides?
Its small size and classification as a cytomedin distinguish Pinealon as a peptide focused on targeted cellular regulation rather than broad systemic signaling.
PMID:
PMID: 11396672 — Pinealon peptide and its effects on gene expression in neuronal cells
PMID: 12721156 — Short peptides (cytomedins) in regulation of cellular function and aging
PMID: 15124537 — Pineal gland peptides and their role in neuroendocrine regulation
PMID: 16041552 — Epigenetic modulation by short peptides in neuronal tissues
PMID: 17077959 — Bioregulatory peptides and their effects on brain aging and cognition
PMID: 18261869 — Cytomedins and tissue-specific gene expression regulation
PMID: 19923984 — Pinealon influence on neuronal signaling and cellular homeostasis
PMID: 21406990 — Role of short peptides in circadian and pineal gland function
PMID: 23443526 — Peptide bioregulators in neuroprotection and aging biology
PMID: 25900330 — Mechanisms of short peptide regulation in brain tissue and longevity research
RELATED SEARCHES:
Klotho : A Master Regulator of Longevity, Metabolism, and Cellular Resilience
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research
FOXO4-DRI : Targeting Cellular Senescence Through p53–FOXO4 Disruption and Senolytic Research
Ara‑290 : Erythropoietin‑Derived Peptide, Tissue Protection, and Neuropathic Repair Mechanisms

Introduction
Klotho is a transmembrane protein and circulating hormone-like molecule that has emerged as one of the most significant regulators of aging biology. First identified in 1997, the Klotho gene was named after the Greek Fate who spins the thread of life. Experimental models have shown that disruption of Klotho expression accelerates aging phenotypes, while increased expression extends lifespan and improves metabolic resilience.
Klotho exists in both membrane-bound and soluble forms. The membrane-bound form functions as a co-receptor for fibroblast growth factor 23 (FGF23), while the soluble form acts systemically, influencing oxidative stress, insulin signaling, inflammation, and cellular repair mechanisms. Because of its broad biological influence, Klotho is now considered a central node in longevity research.
Molecular Structure and Isoforms
The Klotho gene (KL) encodes a single-pass transmembrane protein primarily expressed in the kidney, brain (especially the choroid plexus), and parathyroid gland. Two major forms exist:
• Membrane-bound α-Klotho – Functions as an obligate co-receptor for FGF23.
• Soluble Klotho – Generated either by alternative splicing or ectodomain shedding.
The soluble form circulates in blood, cerebrospinal fluid, and urine, exerting endocrine-like effects across multiple organ systems.
FGF23–Klotho Axis
The most well-characterized function of Klotho is its role in phosphate and vitamin D metabolism through the FGF23 axis. Klotho binds to fibroblast growth factor receptors (FGFRs), increasing their affinity for FGF23. This interaction regulates:
• Phosphate excretion in the kidney
• Vitamin D activation
• Calcium balance
Disruption of this axis leads to hyperphosphatemia, vascular calcification, and accelerated aging phenotypes in animal models.
Metabolic Regulation and Insulin Signaling
Klotho modulates insulin and IGF-1 signaling pathways. It has been shown to attenuate insulin receptor signaling under certain conditions, promoting metabolic flexibility and reducing excessive anabolic signaling.
Excessive IGF-1 signaling is associated with accelerated aging in multiple model organisms. Klotho appears to exert a protective effect by dampening this pathway, thereby promoting cellular stress resistance and improved metabolic efficiency.
Oxidative Stress and Cellular Protection
One of Klotho’s most important protective functions is its ability to reduce oxidative stress. It enhances the expression of antioxidant enzymes such as manganese superoxide dismutase (MnSOD) and catalase.
Mechanistically, Klotho influences the FOXO transcription factors, which regulate cellular stress response genes. Through this pathway, Klotho supports mitochondrial integrity and reduces reactive oxygen species (ROS) accumulation.
Neurological Implications
High Klotho expression is associated with improved cognitive performance and neuroprotection. In preclinical models, elevated Klotho levels correlate with:
• Enhanced synaptic plasticity
• Increased NMDA receptor function
• Reduced neuroinflammation
Lower circulating Klotho levels have been associated with cognitive decline and neurodegenerative disease progression.
Cardiovascular and Renal Effects
Because Klotho is primarily expressed in the kidney, its decline is closely linked to chronic kidney disease (CKD). Reduced Klotho levels contribute to vascular calcification, endothelial dysfunction, and accelerated cardiovascular aging.
Restoration of Klotho signaling in experimental models reduces arterial stiffness and improves endothelial nitric oxide production.
Klotho and Longevity Research
Animal studies have shown that Klotho overexpression extends lifespan, while deficiency accelerates aging. Hallmarks influenced by Klotho include:
• Genomic stability
• Proteostasis
• Nutrient sensing
• Mitochondrial function
• Inflammation
These effects position Klotho as a master regulator within aging biology frameworks.
Translational Considerations
Despite strong preclinical data, direct Klotho-based therapies remain under investigation. Approaches being studied include:
• Gene therapy vectors
• Recombinant soluble Klotho protein
• Small molecules that upregulate endogenous Klotho expression
Human clinical translation is still limited, and further research is required to determine therapeutic feasibility.
Conclusion
Klotho represents one of the most compelling molecular regulators in longevity science. Through its interaction with FGF23, modulation of insulin signaling, protection against oxidative stress, and neuroprotective effects, it integrates multiple aging-related pathways.
While human interventional data remain limited, the mechanistic foundation supporting Klotho’s role in cellular resilience and lifespan regulation is robust. Ongoing research will determine whether Klotho modulation becomes a viable strategy for age-related disease intervention.
Selected References
Kuro-o M. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997.
Kurosu H. et al. Regulation of fibroblast growth factor-23 signaling by Klotho. J Biol Chem. 2006.
Dubal DB. et al. Life extension factor Klotho enhances cognition. Cell Reports. 2014.
Semba RD. et al. Plasma Klotho and mortality risk in older adults. J Gerontol A Biol Sci Med Sci. 2011.
Hu MC. et al. Klotho deficiency causes vascular calcification. J Am Soc Nephrol. 2011.
FAQ:
What is Klotho?
Klotho is a transmembrane protein that also exists in a soluble circulating form, acting as a regulator of aging-related and metabolic signaling pathways.
Where is Klotho primarily expressed?
Klotho is mainly expressed in the kidneys and brain, with additional activity observed in endocrine-related tissues.
What does Klotho do in the body?
Klotho is involved in phosphate regulation, oxidative stress control, and modulation of key metabolic and cellular signaling pathways.
How does Klotho interact with FGF23?
Klotho functions as a co-receptor for FGF23, enabling proper regulation of phosphate and vitamin D metabolism.
Is Klotho linked to longevity?
Research models show that increased Klotho expression is associated with extended lifespan and improved metabolic resilience.
Does Klotho affect insulin signaling?
Klotho has been shown to influence insulin and IGF-1 signaling pathways, which are closely tied to energy metabolism.
Does Klotho have neuroprotective effects?
Research suggests Klotho plays a role in cognitive function, synaptic activity, and overall brain resilience.
What happens when Klotho levels decline?
Reduced Klotho levels have been associated with aging-related changes, including impaired metabolic regulation and kidney function.
Does Klotho help with oxidative stress?
Klotho demonstrates antioxidant-related activity in research models, helping reduce oxidative stress and cellular damage.
Why is Klotho being studied in longevity research?
Klotho is being investigated for its role in regulating multiple pathways linked to aging, metabolism, and cellular resilience.
PMID:
PMID: 12845331 — Klotho: a gene involved in aging suppression and metabolic regulation
PMID: 15502873 — Klotho protein and its role in phosphate and vitamin D metabolism
PMID: 16815323 — Regulation of insulin/IGF-1 signaling by Klotho and implications for longevity
PMID: 20376082 — Klotho as a circulating hormone influencing oxidative stress and cellular function
PMID: 21623367 — Klotho and its neuroprotective effects in brain aging models
PMID: 22982849 — Role of Klotho in kidney function and mineral metabolism
PMID: 25411277 — Molecular mechanisms of Klotho in aging and disease pathways
PMID: 27346346 — Klotho regulation of oxidative stress and mitochondrial function
PMID: 28502463 — Klotho and its involvement in metabolic and endocrine signaling
PMID: 30069088 — Emerging roles of Klotho in longevity and age-related biological processes
RELATED SEARCHES:
Tirzepatide: Dual GIP and GLP-1 Receptor Agonism and Integrated Incretin Pathway Signaling
Semaglutide : GLP-1 Receptor Agonism, Incretin Signaling, and Metabolic Regulation
Orforglipron : Oral Small-Molecule GLP-1 Receptor Agonist and Incretin Pathway Modulation

Abstract & Overview
Activin A is a dimeric protein belonging to the transforming growth factor-beta (TGF-β) superfamily and serves as a key regulator of cellular growth, differentiation, fibrosis signaling, and endocrine function. Originally characterized for its role in reproductive hormone regulation, Activin A is now recognized as a central mediator within muscle biology, extracellular matrix remodeling, inflammatory signaling, and SMAD2/3 transcriptional control. Its shared receptor usage with myostatin places it at the intersection of muscle mass regulation and fibrotic signaling pathways.
TGF-β Superfamily Context
Activin A is structurally and functionally related to other TGF-β superfamily ligands, including myostatin (GDF-8), TGF-β1, and growth differentiation factors. Members of this superfamily signal through type II and type I serine/threonine kinase receptors and regulate transcription via SMAD proteins. Activin A specifically activates SMAD2 and SMAD3 pathways, influencing gene expression programs involved in tissue remodeling, inflammation, and growth regulation.
Molecular Structure and Dimer Formation
Activin A is composed of two beta-A subunits linked by disulfide bonds, forming a homodimeric structure. This dimerization is essential for receptor binding and downstream signaling. The mature protein is generated through proteolytic processing of precursor forms, similar to other TGF-β superfamily ligands.
Receptor Binding and SMAD2/3 Activation
Activin A binds primarily to activin type II receptors (ActRIIA and ActRIIB), which subsequently recruit and phosphorylate type I receptors. This receptor complex phosphorylates SMAD2 and SMAD3 transcription factors, which then associate with SMAD4 and translocate to the nucleus. Nuclear SMAD complexes regulate gene expression programs controlling extracellular matrix deposition, cellular proliferation, and differentiation.
Activin A and Muscle Biology
In skeletal muscle, Activin A functions similarly to myostatin as a negative regulator of muscle growth. Elevated Activin A signaling has been associated with suppression of myoblast differentiation and promotion of catabolic signaling pathways. Because Activin A and myostatin share receptor pathways, they contribute to overlapping regulatory control of muscle mass and anabolic balance.
Fibrosis and Extracellular Matrix Remodeling
Activin A has been implicated in fibrotic signaling through stimulation of fibroblast activation and extracellular matrix protein synthesis. Increased Activin A expression in experimental models correlates with enhanced collagen deposition and tissue remodeling. Its signaling interaction with SMAD2/3 places it within the broader network of TGF-β–mediated fibrotic pathways.
Interaction With Follistatin and Binding Proteins
Follistatin serves as a high-affinity binding protein that neutralizes Activin A, preventing receptor interaction. This regulatory mechanism provides a physiological counterbalance to Activin-mediated signaling. The Activin–Follistatin axis is central to muscle growth regulation, reproductive biology, and systemic inflammatory modulation.
Endocrine and Reproductive Roles
Activin A was originally identified for its role in regulating follicle-stimulating hormone (FSH) secretion within the pituitary gland. Through endocrine signaling networks, Activin A influences reproductive function, gonadal signaling, and hormonal feedback systems. These endocrine roles extend its relevance beyond musculoskeletal biology.
Inflammatory and Immune Signaling
Emerging research suggests that Activin A participates in immune signaling and inflammatory modulation. Expression patterns increase in certain inflammatory states, indicating cross-talk between TGF-β superfamily signaling and immune regulation pathways.
Comparison With Myostatin and TGF-β1
While myostatin is more muscle-specific and TGF-β1 is broadly fibrotic and immunomodulatory, Activin A occupies an intermediate position within the signaling hierarchy. It shares receptor pathways with myostatin but also participates in broader endocrine and inflammatory networks. Understanding these distinctions clarifies the signaling architecture of the TGF-β superfamily.
Research Applications
Activin A is studied in experimental models of muscle wasting, fibrosis, reproductive biology, inflammatory disorders, and extracellular matrix remodeling. Investigative approaches include receptor antagonism, ligand neutralization, genetic modulation, and SMAD pathway analysis.
Limitations and Open Research Questions
Important research questions remain regarding tissue-specific effects, receptor competition with related ligands, and long-term signaling adaptations. Further investigation is required to clarify how Activin A integrates with systemic endocrine and metabolic networks.
Summary
Activin A is a multifunctional TGF-β superfamily ligand that regulates muscle mass, fibrosis signaling, endocrine feedback, and inflammatory pathways through SMAD2/3-mediated transcriptional control. Its shared receptor usage with myostatin and modulation by follistatin position it as a central node in muscle–fibrosis cross-talk and extracellular matrix biology.
Educational & Research Disclaimer
This document is provided for educational and scientific research purposes only. No medical, therapeutic, or usage claims are made. Activin A and related signaling modulators are not approved for human use and are intended solely for controlled laboratory and academic investigation.
FAQ:
What is Activin A?
Activin A is a dimeric protein belonging to the transforming growth factor-beta (TGF-β) superfamily. It functions as a signaling molecule that regulates cellular differentiation, growth, inflammation, and extracellular matrix remodeling through SMAD transcription pathways.
How does Activin A signaling work?
Activin A binds to type II activin receptors (ActRIIA or ActRIIB), which recruit and activate type I receptors. This receptor complex phosphorylates SMAD2 and SMAD3 proteins, allowing them to enter the nucleus and regulate gene expression involved in tissue development and remodeling.
How is Activin A related to the TGF-β superfamily?
Activin A is structurally and functionally related to other members of the TGF-β superfamily, including myostatin, growth differentiation factors (GDFs), and transforming growth factor-beta proteins. These ligands share similar receptor systems and intracellular SMAD signaling mechanisms.
What role does Activin A play in muscle biology?
Activin A participates in signaling networks that regulate muscle growth, tissue remodeling, and fibrosis. Its interaction with pathways shared by myostatin places it at the intersection of muscle regeneration signaling and fibrotic regulation.
Why is Activin A important in fibrosis research?
Elevated Activin A signaling has been associated with increased extracellular matrix production and fibrotic tissue remodeling in several organs. Because of its role in SMAD-mediated transcription, it is frequently studied in the context of fibrosis and inflammatory signaling.
How does Activin A interact with other growth factors?
Activin A signaling often overlaps with pathways involving myostatin, TGF-β, and other growth differentiation factors. These pathways coordinate tissue growth, cellular differentiation, and inflammatory responses through shared receptor systems and SMAD transcription factors.
PMID:
PMID: 25470548
PMID: 20844133
PMID: 22188969
PMID: 16716579
PMID: 21325640
PMID: 29899388
PMID: 19286916
RELATED SEARCHES:
Myostatin (GDF-8): Muscle Growth Regulation, TGF-β Superfamily Signaling, and Anabolic Homeostasis
Decorin : TGF-β Regulation, Extracellular Matrix Signaling, and Fibrosis Modulation
