
Abstract & Overview
Adamax (Ac-MEHFPGPAG-NH₂; C₅₀H₆₉N₁₁O₁₁S; MW 1032.23 g/mol) is a synthetic octapeptide and the most structurally advanced member of the Semax analogue family, a class of neuropeptides derived from the ACTH(4–7) fragment of adrenocorticotropic hormone. Adamax was engineered through two key structural modifications to the parent compound Semax (Met-Glu-His-Phe-Pro-Gly-Pro): N-terminal acetylation, which enhances metabolic stability and membrane permeability, and C-terminal conjugation with an adamantane-based group, which substantially increases lipophilicity, resistance to enzymatic degradation, and blood-brain barrier (BBB) penetration. The compound’s name is a portmanteau of ‘adamantane’ and ‘maximum,’ reflecting the design intent to maximise the pharmacological profile of the Semax scaffold [1][2].
The Semax family of peptides has a well-documented research history originating from the Institute of Molecular Genetics at the Russian Academy of Sciences, where Semax was first described in 1991 as a synthetic analogue of the ACTH(4–7) tetrapeptide fragment Met-Glu-His-Phe. Semax is an approved prescription medication in Russia and Ukraine, where it is used clinically for stroke, transient ischaemic attack, memory and cognitive disorders, optic nerve disease, and immune system support [3][4]. The extensive preclinical and clinical research base established for Semax provides the mechanistic framework from which Adamax’s proposed pharmacological profile is derived, with the adamantane modification anticipated to amplify and extend these effects through improved pharmacokinetic properties [1][5].
“Semax rapidly elevates the levels and expression of brain-derived neurotrophic factor (BDNF) and its signaling receptor tropomyosin receptor kinase B (TrkB) in the hippocampus, and rapidly activates serotonergic and dopaminergic brain systems… it has been found to produce antidepressant-like and anxiolytic-like effects, attenuate the behavioral effects of exposure to chronic stress, and potentiate the locomotor activity produced by D-amphetamine.” — Semax pharmacology, Wikipedia / Dolotov et al. (2006) [5][6].
Adamax is classified as a synthetic nootropic peptide, a cell-penetrating peptide, and a designer analogue of Semax. It has been identified in border seizures and has been submitted for classification as a prescription medicine in New Zealand (Medsafe, 2025). No dedicated peer-reviewed clinical trials have been published specifically for Adamax; its proposed pharmacological profile is derived from the extensive Semax research literature combined with structural pharmacology reasoning regarding the contributions of the adamantane modification. All research applications of Adamax remain strictly preclinical and experimental in nature [1][2].
Molecular Identity and Structural Architecture
Peptide Backbone: The ACTH(4–7) Core and Semax Scaffold
The structural foundation of Adamax is the ACTH(4–7) tetrapeptide fragment Met-Glu-His-Phe (MEHF), which constitutes the biologically active core of the Semax family. This fragment is derived from adrenocorticotropic hormone (ACTH), a 39-amino-acid pituitary peptide, and retains the melanocortin receptor-interacting and neuroprotective properties of the parent hormone without the steroidogenic activity of the full ACTH molecule. In Semax, this tetrapeptide core is extended at the C-terminus with the tripeptide Pro-Gly-Pro (PGP), which confers resistance to enzymatic degradation and contributes additional neuroprotective properties through its own biological activity as a collagen-derived peptide with anti-inflammatory effects [3][4].
Adamax extends the Semax heptapeptide scaffold (MEHFPGP) with two additional residues at the C-terminus (Ala-Gly), yielding the octapeptide sequence MEHFPGPAG. The full Adamax sequence is therefore Ac-Met-Glu-His-Phe-Pro-Gly-Pro-Ala-Gly-NH₂ (Ac-MEHFPGPAG-NH₂). The molecular weight of 1032.23 g/mol reflects the combined contributions of the octapeptide backbone, the N-terminal acetyl group, the C-terminal amide, and the adamantane-based C-terminal modification. The molecular formula C₅₀H₆₉N₁₁O₁₁S includes the single sulfur atom from the methionine residue at position 1 of the sequence [1][2].
The Adamantane Modification: Structure and Pharmacokinetic Rationale
The defining structural feature of Adamax is the adamantane group conjugated to its C-terminus. Adamantane (C₁₀H₁₆) is a tricyclic diamondoid hydrocarbon with a cage-like structure composed of four fused cyclohexane rings in a chair conformation, forming the smallest unit of the diamond crystal lattice. This rigid, symmetrical cage structure confers exceptional lipophilicity, metabolic stability, and three-dimensional bulk that profoundly alters the pharmacokinetic profile of any peptide to which it is conjugated. Adamantane is a well-established pharmacophore in CNS drug design: it is the core structural element of amantadine (Parkinson’s disease, influenza), memantine (Alzheimer’s disease), and rimantadine (influenza), all of which exploit the adamantane cage’s lipophilicity for enhanced CNS penetration [7][8].
In the context of Adamax, the adamantane modification is anticipated to confer three primary pharmacokinetic advantages over unmodified Semax. First, the substantially increased lipophilicity of the adamantane-conjugated peptide is expected to enhance passive diffusion across the blood-brain barrier, increasing CNS bioavailability. Second, the bulky, sterically protected adamantane cage provides resistance to enzymatic degradation by peptidases and enkephalinase, extending the plasma and CNS half-life of the peptide. Third, the N-terminal acetylation, which is present in both N-Acetyl Semax and Adamax, provides additional protection against aminopeptidase-mediated N-terminal degradation, further contributing to metabolic stability. Together, these modifications are designed to produce a peptide with a substantially longer bioactivity window than Semax [1][5][9].
Mechanistic Rationale: Proposed Pathways of Action
BDNF/TrkB Axis: Neurotrophic Signalling and Synaptic Plasticity
The most extensively characterised mechanism through which the Semax family exerts its cognitive and neuroprotective effects is the upregulation of brain-derived neurotrophic factor (BDNF) and its high-affinity receptor tropomyosin receptor kinase B (TrkB) in the hippocampus. BDNF is the most abundant neurotrophin in the adult brain and serves as the master regulator of synaptic plasticity, long-term potentiation (LTP), neurogenesis, and neuronal survival. Its signalling through TrkB activates three major downstream cascades: the PI3K/Akt pathway (promoting neuronal survival and anti-apoptotic signalling), the MAPK/ERK pathway (supporting synaptic plasticity and memory consolidation), and the PLCγ pathway (regulating intracellular calcium and short-term plasticity) [5][6].
Dolotov et al. (2006) demonstrated in rat hippocampus that Semax administration produced a 1.4-fold increase in BDNF protein levels, a 1.6-fold increase in TrkB tyrosine phosphorylation, a 3-fold increase in exon III BDNF mRNA, and a 2-fold increase in TrkB mRNA. These findings established the hippocampal BDNF/TrkB system as a primary mediator of Semax’s cognitive and neuroprotective effects [6]. Adamax, by virtue of its extended half-life and enhanced CNS penetration conferred by the adamantane modification, is proposed to produce a more sustained and potent activation of this same BDNF/TrkB axis. The adamantane group may also enhance TrkB receptor sensitivity in hippocampal and cortical regions, amplifying the neurotrophic signal beyond what is achievable with unmodified Semax [1][9].
Melanocortin Receptor Interactions: MC4R and MC5R
The ACTH(4–7) core of Adamax (Met-Glu-His-Phe) retains the capacity to interact with melanocortin receptors, a family of G protein-coupled receptors (GPCRs) that mediate diverse physiological functions in the CNS. Evidence from Semax research indicates competitive antagonism of α-melanocyte-stimulating hormone (α-MSH) at the MC4 and MC5 receptors in both in vitro and in vivo experimental conditions, suggesting that Semax (and by extension Adamax) may act as an antagonist or partial agonist at these receptor subtypes [3]. MC4R is expressed in the hippocampus, hypothalamus, and cortex, where it plays roles in cognition, energy balance, and stress response. MC5R is expressed in peripheral tissues and the brain, where its functions are less fully characterised. The MC3R may also be a target, though this has not been definitively established [3].
Enkephalinase Inhibition and Endogenous Neuropeptide Preservation
A proposed secondary mechanism of the Semax family involves inhibition of enkephalinase (neutral endopeptidase, neprilysin; EC 3.4.24.11), a zinc-dependent metalloprotease responsible for the degradation of multiple endogenous neuropeptides including enkephalins, substance P, neurotensin, and atrial natriuretic peptide. By inhibiting enkephalinase, Semax and Adamax may increase the synaptic availability of endogenous opioid peptides (enkephalins), contributing to analgesia, mood regulation, and neuroprotection. The adamantane modification in Adamax provides the additional benefit of rendering the peptide itself resistant to enkephalinase-mediated degradation, simultaneously inhibiting the enzyme and protecting the peptide from its activity [3][4].
Neurotransmitter Modulation: Serotonergic, Dopaminergic, and Glutamatergic Systems
Beyond its neurotrophic and receptor-mediated mechanisms, the Semax scaffold exerts broad modulatory effects across multiple neurotransmitter systems. Semax rapidly activates the brain serotonergic system, an effect that has been linked to its anxiolytic and antidepressant properties in animal models. Agapova et al. (2007) demonstrated that chronic Semax administration produced significant anxiolytic and antidepressant effects in rats, attributing these effects to serotonergic activation and hippocampal BDNF upregulation [10]. Dopaminergic modulation has also been documented: Semax augments psychostimulant-induced central dopamine release and potentiates D-amphetamine locomotor activity, suggesting interactions with the mesolimbic and nigrostriatal dopamine systems relevant to motivation, reward, and attention [3][11].
Glutamatergic and GABAergic systems are also implicated in the Semax family’s cognitive effects. The BDNF/TrkB axis directly modulates NMDA receptor function and synaptic AMPA receptor trafficking, both of which are critical for LTP and memory consolidation. Additionally, the adamantane scaffold in Adamax shares structural similarity with memantine, an NMDA receptor antagonist used in Alzheimer’s disease treatment, raising the hypothesis that Adamax may possess additional NMDA receptor modulatory activity beyond what is observed with unmodified Semax. This potential dual mechanism — BDNF/TrkB upregulation combined with NMDA receptor modulation — represents a particularly compelling research hypothesis for Adamax’s cognitive enhancement profile [7][8][9].
HPA Axis Modulation and Stress Resilience
The ACTH(4–7) origin of Adamax’s core sequence establishes a structural connection to the hypothalamic-pituitary-adrenal (HPA) axis, the central neuroendocrine system governing the stress response. While Adamax lacks the steroidogenic activity of full-length ACTH, its ACTH-derived fragment may modulate HPA axis tone through melanocortin receptor interactions in the hypothalamus. Preclinical evidence from Semax research demonstrates that the peptide attenuates the behavioural consequences of chronic stress exposure in animal models, suggesting a stress-resilience mechanism that may be relevant to cognitive performance under adverse conditions. This HPA-modulating property, combined with the BDNF-mediated hippocampal neuroprotection, positions Adamax as a compound of interest for research into stress-induced cognitive impairment [10][11].
Research Applications and Preclinical Evidence
Neuroprotection in Cerebral Ischaemia Models
The most extensively studied application of the Semax family in preclinical research is neuroprotection in models of cerebral ischaemia. Semax has been shown to markedly affect the immune response in rat models of ischaemic brain injury, enhancing the antigen presentation signalling pathway, intensifying interferon signalling, and increasing immunoglobulin heavy chain gene expression. Researchers have proposed that Semax’s neuroprotective mechanism operates through ‘neuroimmune crosstalk,’ with the Pro-Gly-Pro (PGP) component of the peptide playing a key role in coordinating the immune response to ischaemic injury [12]. Semax has also been shown to reduce VEGFA levels after ischaemic brain injury, suggesting an anti-inflammatory mechanism that limits secondary damage [13]. Given Adamax’s enhanced CNS penetration and extended half-life, its neuroprotective potential in ischaemia models represents a primary research hypothesis.
Cognitive Enhancement and Memory Research
Semax’s cognitive-enhancing effects in animal models provide the preclinical foundation for Adamax’s proposed nootropic profile. The peptide has been shown to reduce memory and learning deficits in rats exposed to amphetamines in utero, with researchers concluding that it may enable significant recovery of memory functions in brain-damaged subjects [14]. In glaucoma research, Semax outperformed traditional neuroprotective treatments for glaucomatous optic neuropathy in a 2001 clinical study, demonstrating potent neuroprotective and neurotrophic effects on the visual system [15]. The 2007 ADHD/Rett syndrome hypothesis paper proposed that Semax’s combined augmentation of central dopamine release and BDNF synthesis could be therapeutically relevant in neurodevelopmental disorders characterised by BDNF deficiency and dopaminergic dysregulation [11].
Antioxidant and Heavy Metal Neuroprotection
Beyond ischaemia and cognitive research, the Semax family has demonstrated neuroprotective activity against heavy metal toxicity. Grigoreva et al. (2016) found that Semax counteracted the avoidance response inhibition caused by heavy metal salt poisoning in rats with efficacy comparable to ascorbic acid, confirming antioxidant properties [16]. A separate study demonstrated that Semax reduced copper-induced cytotoxicity in neuronal cells, with researchers noting its neuroprotective activity in the context of metal ion dysregulation relevant to neurodegenerative disorders including Alzheimer’s and Parkinson’s disease [17]. These antioxidant and metal-chelating properties may be further amplified in Adamax through the histidine residue’s known metal-binding capacity and the extended bioavailability conferred by the adamantane modification.
Semax Family Comparative Profile
| Parameter | Semax | N-Acetyl Semax | Adamax |
| Sequence | MEHFPGP | Ac-MEHFPGP | Ac-MEHFPGPAG-NH₂ |
| Molecular Weight | 813.93 g/mol | ~856 g/mol | 1032.23 g/mol |
| N-terminus | Free amine | Acetylated | Acetylated |
| C-terminus | Pro-Gly-Pro-OH | Pro-Gly-Pro-OH | Adamantane-NH₂ |
| Lipophilicity | Moderate | Moderate+ | High |
| BBB Penetration | Moderate | Moderate+ | Enhanced |
| Enzymatic Stability | Moderate | Moderate+ | High |
| BDNF Upregulation | Confirmed (preclinical) | Enhanced (proposed) | Extended (proposed) |
Safety Profile and Regulatory Considerations
No dedicated safety or toxicology studies have been published specifically for Adamax. The parent compound Semax has an established safety profile from decades of clinical use in Russia and Ukraine, where it is administered as a nasal spray at doses of 0.1–1.0 mg/day for neurological conditions, with no significant adverse events reported in the published literature at therapeutic doses. The structural modifications in Adamax — N-terminal acetylation and C-terminal adamantane conjugation — are generally considered to reduce rather than increase toxicological risk, as they primarily affect pharmacokinetic properties (stability, lipophilicity) rather than introducing novel reactive chemical groups. Adamantane itself has a well-established safety profile as the core scaffold of amantadine and memantine, both of which have been used clinically for decades [7][8].
From a regulatory perspective, Adamax has been identified in border seizures in some jurisdictions and has been submitted for classification as a prescription medicine in New Zealand (Medsafe, 2025). It is not approved by the FDA or any major Western regulatory authority as a pharmaceutical agent. Its classification as a designer drug in some jurisdictions reflects regulatory caution regarding novel synthetic peptides rather than confirmed evidence of harm. All research applications of Adamax must be conducted within the applicable regulatory framework of the relevant jurisdiction, and the compound is not appropriate for human use outside of formally approved clinical research settings [1][2].
Conclusion
Adamax represents the most structurally advanced member of the Semax analogue family, combining the well-characterised neuroprotective and cognitive-enhancing scaffold of Semax with two strategic pharmacokinetic enhancements: N-terminal acetylation for aminopeptidase resistance and C-terminal adamantane conjugation for increased lipophilicity, BBB penetration, and enzymatic stability. The compound’s proposed mechanism of action centres on the BDNF/TrkB neurotrophic axis — the same pathway through which Semax’s cognitive and neuroprotective effects have been most rigorously characterised in preclinical models — with the adamantane modification anticipated to produce a more sustained and potent activation of this system. Additional proposed mechanisms include melanocortin receptor (MC4R/MC5R) modulation, enkephalinase inhibition, serotonergic and dopaminergic neurotransmitter modulation, and potentially NMDA receptor interactions analogous to those of the adamantane-containing drug memantine.
The research base for Adamax is currently extrapolated from the extensive Semax literature and structural pharmacology reasoning, as no dedicated peer-reviewed clinical trials for Adamax have been published. The compound’s regulatory classification as a prescription medicine in New Zealand and its identification in border seizures underscore the need for formal preclinical safety and efficacy studies before any clinical research can be conducted. Nevertheless, the convergence of a well-validated neuropeptide scaffold with a pharmacokinetically optimised adamantane modification positions Adamax as a compelling subject for future research in neuroprotection, cognitive enhancement, and stress resilience. Its potential dual mechanism of BDNF upregulation and NMDA modulation, in particular, merits systematic investigation in appropriate preclinical models.
References
[1] Adamax. Wikipedia. https://en.wikipedia.org/wiki/Adamax. Accessed May 2025.
[2] Medsafe New Zealand. Classification of Unscheduled Peptides. Submission to the Medicines Classification Committee. June 2025. https://www.medsafe.govt.nz/
[3] Semax. Wikipedia. https://en.wikipedia.org/wiki/Semax. Accessed May 2025.
[4] Ashmarin IP, Nezavibatko VN, Levitskaya NG, et al. Design and investigation of a nootropic analogue of adrenocorticotropin 4–7 without hormonal activity. Neurosci Behav Physiol. 1997;27(2):188–193. doi:10.1007/BF02462906. PMID: 9109929.
[5] Semaxpolska.com. Adamax Peptide: What It Is, How It Works, Safety, and Scientific Research. https://semaxpolska.com/en/adamax-peptide/. Accessed May 2025.
[6] Dolotov OV, Karpenko EA, Inozemtseva LS, et al. Semax, an analog of ACTH(4–7) with cognitive effects, regulates BDNF and trkB expression in the rat hippocampus. Brain Res. 2006;1117(1):54–60. doi:10.1016/j.brainres.2006.07.108. PMID: 16962080.
[7] Wanka L, Iqbal K, Schreiner PR. The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem Rev. 2013;113(5):3516–3604. doi:10.1021/cr100264t. PMID: 23432396.
[8] Reisberg B, Doody R, Stöffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–1341. doi:10.1056/NEJMoa013128. PMID: 12672860.
[9] APR Health Solutions. Adamax: Comprehensive Guide. Reddit r/APRHealthSolutions. https://www.reddit.com/r/APRHealthSolutions/comments/1q8sndl/adamax_comprehensive_guide/. Accessed May 2025.
[10] Agapova TY, Agniullin YV, Silachev DN, et al. Effects of ACTH(4–7)PGP (Semax) on the behavior of rats in models of depression and anxiety. Zh Vyssh Nerv Deiat Im I P Pavlova. 2007;57(4):422–430. PMID: 17926576.
[11] Kaplan IV, Guseva NV, Nalivaeva NN, Turner AJ. Semax as a potential treatment for ADHD and Rett syndrome. Med Hypotheses. 2007;68(5):1136–1141. doi:10.1016/j.mehy.2006.09.048. PMID: 17126503.
[12] Medvedeva EV, Dmitrieva VG, Povarova OV, et al. The peptide semax affects the expression of genes related to the immune and vascular systems in rat brain with incomplete global ischemia. BMC Neurosci. 2014;15:108. doi:10.1186/1471-2202-15-108. PMC3987924. PMID: 25261150.
[13] Kolomin TA, Shadrina MI, Slominsky PA, et al. A new generation of drugs: synthetic peptides based on natural regulatory peptides. Neurosci Med. 2013;4(4):223–252. doi:10.4236/nm.2013.44033.
[14] Inozemtseva LS, Dolotov OV, Soukhov VV, et al. Semax reduces memory and learning deficits in rat subjects treated with amphetamines in utero. BMC Pharmacol. 2006. PMID: 16822316.
[15] Kaplan IV, Guseva NV, Nalivaeva NN, Turner AJ. Semax for glaucomatous optic neuropathy. 2001. PMID: 14660786.
[16] Grigoreva ME, Manchenko DM, Glazova NY, et al. Semax counteracts heavy metal poisoning in rats. Dokl Biol Sci. 2016;471(1):285–287. doi:10.1134/S0012496616060053. PMID: 28078543.
[17] Grigoreva ME, Manchenko DM, Glazova NY, et al. Semax reduces copper-induced cytotoxicity in neuronal cells. J Inorg Biochem. 2015;145:87–95. doi:10.1016/j.jinorgbio.2014.12.013. PMID: 25862820.
Disclaimer: This article is intended strictly for research and educational review purposes. Adamax is an experimental synthetic peptide that has not been approved by the FDA or any regulatory authority as a pharmaceutical agent. It has been identified in border seizures and is classified as a prescription medicine in New Zealand. No dedicated peer-reviewed clinical trials for Adamax have been published. All proposed mechanisms and effects described in this article are extrapolated from the Semax research literature and structural pharmacology reasoning, and should be treated as hypothetical until confirmed by rigorous preclinical and clinical investigation. This document does not constitute medical advice, endorsement of any compound, or guidance for personal use.
thepeptidecompany.xyz | Research Division
What is Adamax primarily studied for?
Adamax is studied for its interaction with mitochondrial energy pathways, oxidative metabolism, and cellular performance signaling in experimental models.
How does Adamax relate to mitochondrial research?
Research models investigate Adamax for its potential influence on mitochondrial efficiency, ATP production, and oxidative phosphorylation pathways.
What biological pathways are associated with Adamax?
It is commonly studied in pathways involving cellular energy regulation, metabolic flexibility, endurance-associated signaling, and mitochondrial respiration.
Why is Adamax linked to endurance-related research?
Experimental studies explore its association with energy utilization and oxidative metabolism pathways involved in sustained cellular performance.
Is Adamax a peptide or small molecule compound?
Adamax is generally categorized as a research peptide investigated for metabolic and mitochondrial signaling applications.
What research applications commonly involve Adamax?
Adamax is frequently researched in laboratory models focused on mitochondrial bioenergetics, exercise-associated signaling, metabolic stress adaptation, and cellular energy production.
PMID:
31253884 — Mitochondrial bioenergetics and metabolic signaling pathways
29923263 — Oxidative phosphorylation and cellular energy regulation
28446474 — Endurance-associated metabolic adaptation research
31501082 — Mitochondrial efficiency and ATP production mechanisms
26780211 — Skeletal muscle energy metabolism studies
34140407 — Cellular respiration and oxidative metabolism pathways
25609842 — Exercise-associated mitochondrial signaling research
32669311 — Metabolic flexibility and mitochondrial adaptation studies

Adamax 5mg
Adamax is a research peptide studied for its interaction with mitochondrial function, cellular energy pathways, and exercise-associated metabolic signaling in experimental models. It is commonly investigated in endurance-related research, oxidative metabolism, and energy regulation studies.
RELATED SEARCHES:
Semax : ACTH(4–10)-Derived Heptapeptide and Neurotrophic Research Pathways
Noopept : Neuropeptide Derived
Dihexa — Neurotrophic Peptide Research Article (Educational • Research Use Only)
Cerebrolysin : Neurotrophic Peptide
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research

Abstract & Overview
Delta Sleep-Inducing Peptide (DSIP) is an endogenous nonapeptide originally isolated in 1974 from the cerebral venous blood of rabbits during an induced state of sleep. Despite its name, decades of subsequent research have revealed that DSIP is far more than a simple somnogenic agent. It acts as a profound, multifaceted neuroendocrine modulator capable of regulating the hypothalamic-pituitary-adrenal (HPA) axis, altering neurotransmitter dynamics, and exerting significant stress-limiting and neuroprotective effects [1] [2].
While its precise genetic precursor and definitive receptor remain elusive, DSIP’s broad distribution across the hypothalamus, limbic system, and pituitary gland points to its fundamental role in maintaining physiological homeostasis. In experimental models, DSIP has demonstrated remarkable efficacy in promoting slow-wave sleep (SWS), attenuating stress-induced cortisol release, modulating the release of luteinizing hormone (LH) and growth hormone (GH), and even providing anticonvulsant and analgesic properties [3] [4].
“DSIP is an amphiphilic peptide that co-localizes with many peptide and non-peptide mediators in the pituitary and gut. Its ability to interact with the MAPK cascade and its homology to glucocorticoid-induced leucine zipper (GILZ) suggest it serves as a critical link between circadian mechanisms, stress adaptation, and neuroendocrine regulation.” — Gimble et al., Obesity Reviews [5].
Molecular Identity and Structural Architecture
DSIP is a highly conserved, amphiphilic nonapeptide with a molecular weight of 850 Daltons. Its specific amino acid sequence is Trp-Ala-Gly-Gly-Asp-Ala-Ser-Gly-Glu (WAGGDASGE). The structure is unique in that it lacks a defined genetic origin in mammalian models; however, BLAST alignments suggest homology with certain bacterial proteins, hinting at a highly conserved evolutionary lineage [6].
In vivo, native DSIP has a relatively short half-life of approximately 15 minutes due to rapid degradation by aminopeptidase-like enzymes [7]. To counteract this, endogenous DSIP is believed to complex with specific carrier proteins. In clinical and research settings, synthetic analogues and specialized preparations (such as Deltaran) are often utilized to enhance molecular stability and prolong bioactivity [8].
Mechanistic Rationale: CNS and Endocrine Signaling
The pharmacological utility of DSIP lies in its broad, systemic modulatory capacity. Rather than acting as a direct agonist for a single receptor, DSIP functions as a regulatory peptide that restores homeostasis across multiple neural and endocrine networks.
Neurotransmitter Modulation and Sleep Architecture
The primary action of DSIP in the central nervous system involves the induction of spindle and delta EEG activity, the hallmark of deep, restorative slow-wave sleep (SWS) [1]. Recent studies utilizing DSIP fusion peptides (such as DSIP-CBBBP) in PCPA-induced insomnia models have demonstrated its ability to significantly modulate and restore neurotransmitter balance. DSIP administration regulates levels of serotonin (5-HT), dopamine (DA), glutamate, and melatonin, effectively reversing neurotransmitter dysregulation associated with severe sleep deprivation [9].
HPA Axis Regulation and Stress Limitation
DSIP exerts a powerful inhibitory effect on the stress response. Research indicates that DSIP significantly reduces corticotropin-releasing factor (CRF)-induced corticosterone release at the level of the pituitary gland [10]. By decreasing basal corticotropin (ACTH) levels and blocking its stress-induced release, DSIP acts as a potent stress-limiting factor. Furthermore, its homology to GILZ (glucocorticoid-induced leucine zipper) allows it to interact with the MAPK cascade, preventing Raf-1 activation and inhibiting ERK phosphorylation—a crucial pathway in cellular stress signaling [5].
Pituitary Hormone Release: LH and GH
Beyond the HPA axis, DSIP modulates the hypothalamic-pituitary-gonadal (HPG) and somatotropic axes. In rat models, microinjections of DSIP have been shown to stimulate the release of luteinizing hormone (LH) [11]. Additionally, DSIP is a physiological stimulus for sleep-related growth hormone (GH) release. Sleep deprivation naturally increases endogenous DSIP, which in turn drives the secretion of somatoliberin (GHRH) and GH while inhibiting somatostatin [3].
Research Applications and Experimental Evidence
Substance Withdrawal and Addiction Recovery
One of the most compelling clinical applications of DSIP involves the treatment of opioid and alcohol withdrawal syndromes. DSIP has been shown to act antagonistically on opiate receptors, significantly inhibiting the development of dependence. In landmark clinical trials, administration of DSIP produced a beneficial, immediate-onset alleviation of withdrawal symptoms in 97% of opiate-dependent and 87% of alcohol-dependent patients, drastically reducing anxiety and sleep disturbances during detoxification [12] [13].
Mitochondrial Protection and Antioxidant Effects
In vitro studies on rat brain mitochondria have revealed that DSIP enhances the efficiency of oxidative phosphorylation. Under conditions of experimental hypoxia, DSIP demonstrated pronounced stress-protective and antioxidant properties, preserving mitochondrial respiratory chain function and mitigating cellular damage [14].
Narcolepsy and Sleep Disorders
While initially studied for insomnia, DSIP has shown paradoxical benefits in narcolepsy. In clinical case studies, DSIP administration reduced the frequency of daytime sleep attacks while simultaneously increasing daytime activity, alertness, and performance. The nocturnal sleep period was compressed with fewer interruptions, suggesting that DSIP normalizes the overall circadian rhythm rather than simply acting as a sedative [15].
Systemic Effects of DSIP Administration
| Physiological System | Observed Effects in Research Models |
| Central Nervous System | Induces delta EEG activity (SWS); restores 5-HT, DA, and glutamate balance; provides anticonvulsant and analgesic effects. |
| HPA Axis (Stress) | Reduces CRF-induced corticosterone release; lowers basal ACTH; modulates MAPK/ERK cellular stress signaling. |
| Endocrine System | Stimulates LH release; promotes sleep-related GH secretion; inhibits somatostatin. |
| Mitochondrial Function | Enhances oxidative phosphorylation efficiency; provides antioxidant protection during hypoxia. |
| Receptor Interaction | Modulates NMDA receptors; acts antagonistically on opiate receptors to alleviate withdrawal symptoms. |
Conclusion
Delta Sleep-Inducing Peptide represents a highly versatile and potent endogenous regulatory compound. Far surpassing its initial designation as a sleep-promoting agent, DSIP serves as a crucial neuroendocrine modulator that bridges the gap between circadian regulation, stress adaptation, and hormonal balance. Its proven efficacy in restoring slow-wave sleep, blunting cortisol responses, protecting mitochondrial function, and alleviating severe withdrawal syndromes underscores its immense value in both neurological and endocrine research. As investigations into stable synthetic analogues continue, DSIP remains one of the most promising peptides for comprehensive CNS and metabolic restoration.
References
[1] Schoenenberger GA, et al. A naturally occurring delta-EEG enhancing nonapeptide in rabbits. European Journal of Physiology. 1977;369(2):99-109.
[2] Kovalzon VM, Strekalova TV. Delta sleep-inducing peptide (DSIP): a still unresolved riddle. Journal of Neurochemistry. 2006;97(2):303-309.
[3] Iyer KS, Marks GA, Kastin AJ, McCann SM. Evidence for a role of delta sleep-inducing peptide in slow-wave sleep and sleep-related growth hormone release in the rat. PNAS. 1988;85(10):3653-3656.
[4] Schoenenberger GA. Characterization, properties and multivariate functions of Delta-Sleep Inducing Peptide (DSIP). European Neurology. 1984;23(5):321-345.
[5] Gimble JM, et al. Delta sleep-inducing peptide and glucocorticoid-induced leucine zipper: potential links between circadian mechanisms and obesity? Obesity Reviews. 2009;10:46-51.
[6] Delta-sleep-inducing peptide. Wikipedia. https://en.wikipedia.org/wiki/Delta-sleep-inducing_peptide
[7] Pollard BJ, Pomfrett CJ. Delta sleep-inducing peptide. European Journal of Anaesthesiology. 2001;18(7):419-422.
[8] Khvatova EM, et al. Delta sleep inducing peptide (DSIP): effect on respiration activity in rat brain mitochondria and stress protective potency under experimental hypoxia. Peptides. 2003;24(2):307-311.
[9] Mu X, et al. Pichia pastoris secreted peptides crossing the blood-brain barrier and DSIP fusion peptide efficacy in PCPA-induced insomnia mouse models. Frontiers in Pharmacology. 2024;15:1439536.
[10] Graf MV, Kastin AJ, Coy DH, Fischman AJ. Delta-Sleep-Inducing Peptide Reduces CRF-Induced Corticosterone Release. Neuroendocrinology. 1985;41(4):353-356.
[11] Iyer KS, McCann SM. Delta sleep inducing peptide (DSIP) stimulates the release of LH but not FSH via a hypothalamic site of action in the rat. Brain Research Bulletin. 1987;19(5):535-538.
[12] Dick P, Costa C. DSIP in the treatment of withdrawal syndromes from alcohol and opiates. European Neurology. 1984;23(5):364-371.
[13] Kastin AJ, et al. Opioid detoxification with delta sleep-inducing peptide: results of an open clinical trial. Journal of Clinical Psychopharmacology. 1998.
[14] Bobyntsev II, et al. Influence of delta-sleep peptide on the enzymatic activity of the mitochondrial electron transport chain in various rat tissues with aging of the organism. Advances in Gerontology. 2015.
[15] Schneider-Helmert D. Effects of DSIP on narcolepsy. European Neurology. 1984;23(5):353-364.
Disclaimer: This article is intended strictly for research and educational review purposes. The compounds discussed (DSIP) are for laboratory research use only and should only be handled by qualified professionals. This document does not constitute medical advice, nor should it be used to guide clinical practice or personal health decisions.
FAQ:
What is DSIP (Delta Sleep-Inducing Peptide)?
DSIP is a naturally occurring neuropeptide studied for its association with sleep regulation and neuroendocrine signaling.
What is DSIP primarily researched for?
It is studied for its potential role in sleep architecture, circadian rhythm modulation, and stress-response pathways in experimental models.
How does DSIP interact with the nervous system?
DSIP is associated with central nervous system activity and may influence neurotransmitter balance and hypothalamic regulation.
What pathways are linked to DSIP?
Research explores its involvement in circadian rhythm signaling, hormonal regulation, stress adaptation, and sleep-cycle modulation.
Is DSIP only related to sleep?
No, it is also studied for broader neuroendocrine effects, including interactions with cortisol, growth hormone, and stress-related pathways.
Why is DSIP studied in research models?
It provides insight into mechanisms of sleep regulation, neuropeptide signaling, and the relationship between stress and recovery.
PMID:
6992279 — Isolation and characterization of DSIP
7406237 — DSIP and sleep regulation studies
6186123 — DSIP effects on circadian rhythms
6307659 — Neuroendocrine activity of DSIP
6609105 — DSIP and stress-response mechanisms
6966726 — DSIP influence on hormonal regulation
7284026 — Central nervous system effects of DSIP
10362645 — DSIP and sleep-cycle modulation
RELATED SEARCHES:
GHRP‑2 : Pituitary Axis Modulation, Ghrelin Receptor Activation, and Cellular Recovery Research

DSIP 5mg
Research-grade DSIP (Delta Sleep-Inducing Peptide) supplied as a 5mg lyophilized peptide vial for controlled laboratory and in-vitro neurobiology and sleep studies. For research use only.
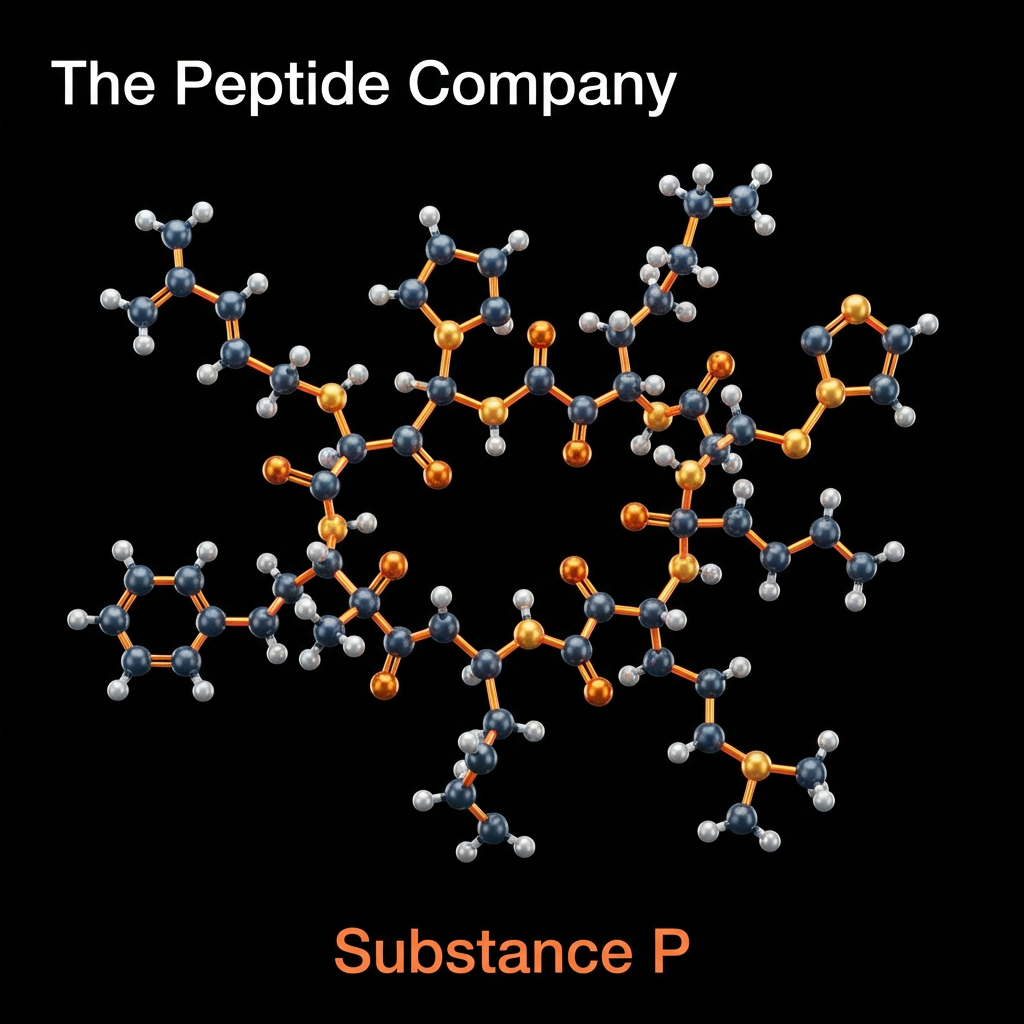
Substance P (SP) is an 11-amino acid neuropeptide belonging to the tachykinin family, widely distributed throughout the central and peripheral nervous systems. First identified in 1931 by von Euler and Gaddum, it holds the distinction of being the first neuropeptide to be discovered. In contemporary research, Substance P is extensively studied as a primary mediator of nociception (pain transmission) and neurogenic inflammation. It functions principally through high-affinity binding to the Neurokinin-1 (NK1) receptor, a G-protein coupled receptor (GPCR) found on neurons, immune cells, and endothelial tissues.
The peptide sequence of Substance P (Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-NH2) is highly conserved across mammalian species, reflecting its critical evolutionary role in survival mechanisms. Beyond its classical function as a neurotransmitter of pain signals in the dorsal horn of the spinal cord, Substance P is now recognized as a ubiquitous modulator of physiological stress responses, immune system regulation, and emotional behavior. Research models involving NK1 receptor antagonism have provided profound insights into the peptide’s involvement in conditions ranging from chronic pain and inflammatory diseases to depression and anxiety disorders.
MOLECULAR STRUCTURE AND NEURO KININ RECEPTOR FAMILY
Substance P is encoded by the preprotachykinin-A (TAC1) gene and is synthesized as a larger precursor protein that undergoes post-translational cleavage. It belongs to the tachykinin peptide family, which also includes Neurokinin A and Neurokinin B. These peptides share a common C-terminal sequence, Phe-X-Gly-Leu-Met-NH2, which is essential for receptor activation. However, Substance P exhibits unique selectivity for the NK1 receptor, distinguishing its biological profile from other family members that prefer NK2 or NK3 receptors.
Structural biology research has elucidated that the C-terminal region of Substance P is responsible for receptor binding and activation, while the N-terminal sequence determines metabolic stability and specificity. The amidation of the C-terminus is critical for biological potency; non-amidated analogs show drastically reduced affinity for the NK1 receptor. This structure-activity relationship is a key focus in the development of synthetic NK1 antagonists designed to block Substance P signaling without interfering with other tachykinin pathways.
The most well-characterized role of Substance P is its function as a nociceptive neurotransmitter. It is synthesized in the cell bodies of dorsal root ganglion (DRG) neurons and transported to both central and peripheral nerve terminals. In response to noxious stimuli—such as intense heat, mechanical pressure, or chemical irritants—Substance P is released into the dorsal horn of the spinal cord, where it acts synergistically with glutamate to transmit pain signals to the brain.
Research into chronic pain models has demonstrated that inhibiting Substance P signaling can attenuate central sensitization, the process by which the nervous system becomes hypersensitive to stimuli. This has led to extensive investigation of NK1 receptor antagonists as potential analgesics. Although early clinical trials faced challenges due to species-specific differences in receptor pharmacology, preclinical data continues to support the critical role of Substance P in the maintenance of neuropathic and inflammatory pain.
NEUROIMMUNE INTERACTIONS AND INFLAMMATORY REGULATION
Substance P serves as a vital bridge between the nervous and immune systems. NK1 receptors are expressed on a wide array of immune cells, including macrophages, mast cells, T-lymphocytes, and dendritic cells. The release of Substance P from peripheral nerve endings can directly activate these cells, triggering the release of pro-inflammatory cytokines like IL-1β, IL-6, and TNF-α. This bi-directional communication is fundamental to the concept of neurogenic inflammation.
This neuroimmune axis is particularly relevant in research on stress-induced immune suppression and inflammatory disorders. By modulating cytokine profiles, Substance P can influence the progression of inflammatory responses. Experimental models have shown that NK1 receptor blockade can reduce the severity of inflammation in conditions like colitis and arthritis, highlighting the therapeutic potential of targeting this neuropeptide in autoimmune pathology.
NEUROGENIC INFLAMMATION AND WOUND HEALING RESEARCH
Neurogenic inflammation refers to the process where inflammatory symptoms—redness, swelling, and heat—are initiated by the release of neuropeptides from sensory nerves rather than by immune cells directly. Substance P is the primary mediator of this process. Upon release from peripheral nerve terminals, it causes vasodilation and plasma protein extravasation (leakage of fluid from blood vessels) by acting on endothelial cells.
Research in regenerative medicine is exploring the dual nature of Substance P. While its pro-inflammatory effects can be detrimental in chronic disease, its ability to stimulate cellular proliferation and angiogenesis is beneficial for tissue repair. Investigational therapies aim to harness its wound-healing properties—particularly in neuropathic ulcers and corneal injuries—while managing the inflammatory component.
SUBSTANCE P IN NEUROPSYCHIATRIC AND STRESS RESPONSE MODELS
Beyond the spinal cord and periphery, Substance P is highly concentrated in brain regions associated with emotional processing, such as the amygdala, hypothalamus, and hippocampus. It is a key component of the stress response system. Animal models of stress and anxiety consistently show elevated release of Substance P in these limbic areas, suggesting it functions as a “stress neurotransmitter.”
Although clinical translation for depression has been mixed, research
continues to investigate Substance P antagonists for specific stress-related disorders, including PTSD and alcohol dependence. The hypothesis is that blocking NK1 signaling may dampen the overactive stress circuitry without the sedative side effects common to benzodiazepines.
PHARMACOLOGICAL MODULATION: NK1 RECEPTOR ANTAGONISTS
The broad physiological role of Substance P has driven the development of NK1 receptor antagonists. The most successful clinical application to date is aprepitant, used to prevent chemotherapy-induced nausea and vomiting (CINV). Substance P is a major mediator of the vomiting reflex in the brainstem, and blocking its receptor effectively suppresses this response.
SOURCEDSTUDIES
- (1)Steinhoff, M.S., et al. “The role of neurokinin-1 receptors in physiology and pathophysiology.” PhysiologicalReviews, vol. 94, no. 1, 2014, pp. 265-301. DOI: 10.1152/physrev.00040.2012.
- (2)Zieglgänsberger, W. “Substance P and pain chronicity.” CellandTissueResearch, vol. 375, no. 1, 2019, pp. 227-241. DOI: 10.1007/s00441-018-2922-y.
- (3)Mashaghi, A., et al. “Neuropeptide substance P and the immune response.” CellularandMolecular LifeSciences, vol. 73, no. 22, 2016, pp. 4249-4264. DOI: 10.1007/s00018-016-2293-z.
- (4)Suvas, S. “Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis.” Journal ofImmunology, vol. 199, no. 5, 2017, pp. 1543-1552. DOI: 10.4049/jimmunol.1601751.
- (5)Ebner, K., et al. “Substance P in stress and anxiety: behavioral changes, mechanisms, and novel therapeutic targets.” CurrentOpinioninPharmacology, vol. 6, no. 5, 2006, pp. 475-481. DOI: 10.1016/j.coph.2006.05.004.
- (6)Navari, R.M., et al. “Neurokinin-1 receptor antagonists in the treatment of chemotherapy-induced nausea and vomiting.” DrugDesign,DevelopmentandTherapy, vol. 10, 2016, pp. 2567-2575. DOI: 10.2147/DDDT.S109404.
FAQ:
What is Substance P?
Substance P is an 11-amino acid neuropeptide belonging to the tachykinin family, widely distributed throughout the central and peripheral nervous systems.
What receptor does Substance P primarily interact with?
Substance P primarily binds to the neurokinin-1 (NK1) receptor, a G-protein coupled receptor expressed on neurons, immune cells, and endothelial tissues.
What biological processes is Substance P associated with?
It is studied for its role in nociceptive transmission, neurogenic inflammation, neuroimmune signaling, and sensory neuron communication.
Where is Substance P found in the body?
Substance P is present in the brain, spinal cord, peripheral sensory neurons, and various immune-related tissues.
How does Substance P relate to pain signaling?
It is widely researched as a neurotransmitter involved in transmitting nociceptive signals between peripheral nerves and central nervous system pathways.
Is Substance P involved in inflammation pathways?
Research indicates Substance P participates in neurogenic inflammation through interactions with immune cells and vascular signaling.
What type of peptide is Substance P?
Substance P is a tachykinin neuropeptide composed of 11 amino acids with conserved structure across mammalian species.
Why is Substance P studied in neuroimmune research?
It is investigated for its role in communication between the nervous system and immune signaling pathways.
Does Substance P act in both central and peripheral systems?
Yes, Substance P is studied in both central nervous system signaling and peripheral sensory neuron pathways.
PMID:
PMID: 7681072 — Distribution and function of Substance P in the nervous system
PMID: 7542593 — Neurokinin-1 receptor binding and Substance P signaling
PMID: 1374954 — Substance P and nociceptive transmission mechanisms
PMID: 1714860 — Tachykinins and neurogenic inflammation pathways
PMID: 7521983 — Substance P in neuroimmune communication
PMID: 1693883 — Sensory neuron release of Substance P
PMID: 8381513 — NK1 receptor pharmacology and signaling
PMID: 10844083 — Substance P in central nervous system signaling
PMID: 11877335 — Substance P and inflammatory mediator pathways
PMID: 15265877 — Neurokinin receptors and peptide neurotransmission
RELATED SEARCHES:
Cerebrolysin : Neurotrophic Peptide
Noopept : Neuropeptide Derived
Thymalin: Thymic Bioregulator Peptide, Immune Aging, and Epigenetic Control of Cellular Homeostasis
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research

MixtureResearch, BDNFandNGF Signaling, and Neuroprotective Mechanisms in Neurological Models
Cerebrolysin represents a highly sophisticated and extensively researched neurotrophic peptide mixture utilized in advanced neurological recovery models. The historical development of this unique compound traces back several decades, originating from an intricate process of extracting and purifying brain tissue. Specifically, the formulation is derived from purified porcine brain proteins, which are carefully processed to create a stable, biologically active therapeutic agent. Over the years, the scientific community has heavily scrutinized this preparation, transitioning its status from a traditional biological extract to a highly defined, multi component polypeptide bioregulator capable of exerting profound disease modifying effects within the central nervous system.
In pharmacological terms, Cerebrolysin is classified as a complex peptide mixture rather than a single synthetic molecule. This classification is vital to understanding its broad spectrum mechanism of action. Unlike modern synthetic drugs that typically target a single receptor or isolated enzymatic pathway, this biological preparation mimics the highly complex, synergistic signaling environments naturally found within a healthy mammalian brain. The diverse array of small peptides operates collectively to modulate entire networks of cellular survival, structural remodeling, and metabolic efficiency, establishing a pharmacological profile that is notoriously difficult to replicate with isolated synthetic compounds.
The manufacturing process of Cerebrolysin relies on standardized enzymatic hydrolysis, a highly controlled biochemical technique designed to break down massive, immunogenic porcine brain proteins into tiny, safe, and biologically active fragments. By utilizing specific proteolytic enzymes under strict laboratory conditions, researchers ensure that the resulting peptide mixture is devoid of large proteins, lipids, and antigenic cellular debris. This meticulous enzymatic cleavage
generates a consistent pool of low molecular weight peptides that can
safely navigate systemic circulation and ultimately penetrate the highly restrictive protective barriers of the human brain.
Today, the neurotrophic and neuroprotective research applications surrounding Cerebrolysin are vast and encompass some of the most challenging conditions in modern neurology. Experimental models heavily utilize this peptide complex to investigate neuronal survival following acute ischemic stroke, progressive neurodegeneration in Alzheimer’s disease, diffuse axonal injury in severe traumatic brain injury, and chronic cognitive decline associated with vascular dementia. By evaluating how this mixture influences the brain at a fundamental molecular level, scientists continue to uncover the profound regenerative capabilities of natural neurotrophic signaling.
COMPOSITIONANDMOLECULARCHARACTERISTICS
The biochemical composition of Cerebrolysin is highly distinctive and serves as the foundation for its pleiotropic therapeutic effects. The rigorous enzymatic hydrolysis process yields a final solution characterized primarily by an abundance of very low molecular weight peptides. Analytical laboratory techniques consistently confirm that the vast majority of the active peptide fragments within the formulation are smaller than 10 kilodaltons. This specific molecular weight distribution is an absolute prerequisite for central nervous system research, as larger protein molecules are categorically excluded from entering the brain parenchyma.
The free amino acid content, which constitutes a significant portion of the total dry weight, includes vital neuroactive building blocks such as glutamate, aspartate, and glycine. However, the primary pharmacological
activity is attributed to the active neuropeptide fractions. These small
peptide sequences act as direct molecular mimics of naturally occurring human survival factors. By maintaining such a low molecular weight profile, the active constituents easily bypass the formidable obstacles that typically hinder neurological drug delivery, ensuring that the active signaling molecules reach their intended targets within the cerebral cortex and hippocampus.
This molecular weight distribution distinctly separates Cerebrolysin from synthetic single peptides. While a synthetic peptide contains only one specific amino acid sequence designed for one specific target, the complex mixture approach ensures that multiple cellular receptors are activated simultaneously. This multi targeted biological strategy effectively prevents the cellular desensitization and feedback inhibition frequently observed in single molecule experimental pharmacology.
BDNF,NGF,CNTF,ANDNT-3MIMETICACTIVITY
The most extensively researched aspect of Cerebrolysin is its ability to directly mimic the action of multiple endogenous neurotrophic factors simultaneously. Neurotrophins are specialized proteins that regulate the growth, survival, and functional maintenance of neurons. By presenting a complex matrix of peptide analogs, Cerebrolysin essentially functions as a broad spectrum neurotrophic substitute, capable of stimulating numerous survival pathways that are typically compromised during severe neurological disease or acute brain injury.
In addition to mimicking brain derived neurotrophic factor, researchers have documented profound nerve growth factor mimetic effects. Nerve growth factor is absolutely critical for the survival of cholinergic neurons, which are heavily implicated in memory formation and cognitive processing. Cerebrolysin contains peptide sequences that successfully bind and activate the TrkA receptor, replicating the natural survival signals required by these vulnerable cholinergic networks. This specific interaction is a primary focus in laboratory models of progressive memory loss.
This simultaneous, synergistic activation is why the peptide mixture is so heavily favored in advanced research. The brain relies on a highly coordinated symphony of growth factors to maintain synaptic health. By delivering a mixture that engages TrkA, TrkB, and other neurotrophic receptors concurrently, researchers can successfully replicate the complex biological conditions necessary for profound tissue regeneration and neurological repair.
NEUROPROTECTIONINISCHEMICSTROKEMODELS
During an acute ischemic stroke, the sudden interruption of cerebral blood flow deprives brain tissue of oxygen and glucose, initiating a catastrophic biochemical cascade known as glutamate excitotoxicity. As energy reserves deplete, dying neurons release massive quantities of glutamate, which hyperactivates neighboring cellular receptors and causes widespread tissue destruction. Cerebrolysin has been exhaustively studied for its ability to interrupt this specific pathological cascade and
preserve the vulnerable tissue surrounding the primary stroke core,
known as the ischemic penumbra.
Beyond modulating excitatory receptors, the peptide complex exerts powerful protective effects on cellular energy centers. Ischemic conditions cause damaged mitochondria to leak highly reactive oxidative molecules. Research confirms that Cerebrolysin administration promotes profound free radical scavenging and mitochondrial protection, stabilizing the mitochondrial membrane and preventing the secondary oxidative damage that normally destroys fragile cellular structures during the reperfusion phase of a stroke.
These clinical stroke trial data points heavily emphasize the value of multi targeted neuroprotection. Because a stroke triggers inflammation, oxidative stress, and excitotoxicity simultaneously, a compound like Cerebrolysin that can address all three pathological avenues concurrently provides a highly effective therapeutic strategy for acute neurovascular emergencies.
ALZHEIMER’SDISEASEANDAMYLOIDPATHOLOGYRESEARCH
The progressive cognitive decline observed in Alzheimer’s disease is pathologically characterized by the accumulation of toxic amyloid beta
plaques and the formation of intracellular neurofibrillary tangles
composed of hyperphosphorylated tau proteins. Research models utilize Cerebrolysin to investigate how neurotrophic signaling can alter the fundamental progression of these toxic protein aggregations and preserve the specific neuronal populations most vulnerable to the disease.
One of the most critical aspects of Alzheimer’s disease research involves the preservation of the basal forebrain. The cholinergic neurons in this region are responsible for acetylcholine synthesis, a neurotransmitter entirely necessary for memory formation. Because these specific neurons are highly dependent on nerve growth factor for their survival, the targeted NGF mimetic effects of Cerebrolysin provide highly specific cholinergic neuron preservation, protecting the brain’s memory centers from toxic amyloid exposure.
By actively reducing the formation of toxic protein aggregates while simultaneously boosting the survival signaling of vulnerable memory networks, Cerebrolysin represents a comprehensive approach to neurodegenerative disease research, highlighting the necessity of combining structural protection with functional neurotransmitter support.
NEUROGENESISANDSYNAPTICPLASTICITYMECHANISMS
The adult mammalian brain retains a remarkable ability to adapt, rewire, and generate new neurons in response to learning and environmental stimuli, a phenomenon known as neuroplasticity. Cerebrolysin is heavily utilized in research environments to study how exogenous peptides can artificially amplify these natural mechanisms of synaptic plasticity and accelerate structural brain remodeling following trauma or during natural aging.
At the molecular level, the enhancement of memory and learning requires structural changes at the synapse. The neurotrophic signals provided by Cerebrolysin travel from the cell surface to the nucleus, where they initiate profound changes in gene expression. The primary driver of this process is the cyclic AMP response element binding protein. Through robust CREB phosphorylation, the peptide mixture triggers the transcription of genes that physically build new synaptic connections and reinforce existing neural circuits.
Beyond creating entirely new neurons, the mixture highly stimulates axonal sprouting and synaptogenesis data in damaged regions. When compared to single factor neurotrophins in plasticity models, the complex mixture consistently yields superior structural remodeling. This suggests that the simultaneous activation of multiple distinct growth factor receptors is required to optimize the highly energy intensive process of building and maintaining new cellular architecture in the central nervous system.
TRAUMATICBRAININJURYANDNEUROREHABILITATIONRESEARCH
Traumatic brain injury initiates a complex, highly destructive secondary injury cascade that can persist for months or years following the initial
mechanical impact. The sheer physical force of the trauma causes diffuse
axonal injury, stretching and tearing delicate nerve fibers and disrupting essential communication networks throughout the brain. Cerebrolysin has emerged as a premier research compound for investigating how to halt this secondary damage and accelerate advanced neurorehabilitation.
In addition to cognitive parameters, severe brain trauma typically results in profound motor deficits. Laboratory research shows that the neurotrophic support provided by the peptide mixture actively facilitates motor function restoration by promoting the rerouting of motor pathways around areas of necrotic tissue, a process heavily reliant on the peptide’s ability to stimulate local axonal sprouting.
This research clearly illustrates that providing the brain with abundant neurotrophic resources during the critical post injury window can drastically alter the final degree of permanent neurological disability, emphasizing the importance of early biochemical intervention following physical brain trauma.
ANTI-INFLAMMATORYANDANTI-APOPTOTICMECHANISMS
Chronic neuroinflammation and programmed cell death, or apoptosis, are common pathological denominators across virtually all major neurological disorders. Whenever brain tissue is damaged, specialized
immune cells called microglia become activated, releasing a storm of
toxic inflammatory cytokines that inadvertently destroy healthy surrounding neurons. Cerebrolysin research heavily focuses on its capacity to act as a powerful neuro immune modulator, calming this destructive inflammatory response while triggering internal cellular survival programs.
In tandem with its anti inflammatory properties, the peptide mixture exerts direct control over the delicate internal mechanisms of apoptosis. When a neuron experiences severe metabolic stress, it weighs pro survival signals against pro death signals. The neurotrophic components of Cerebrolysin heavily tip this scale in favor of survival by altering the expression of specific mitochondrial proteins.
This dual mechanism approach, combining powerful external inflammatory suppression with potent internal apoptotic inhibition, represents the pinnacle of modern neuroprotective strategy, showcasing the immense biological efficiency of complex peptide bioregulators.
COMPARATIVEANALYSISANDTRANSLATIONAL RESEARCH CONSIDERATIONS
As the field of advanced neurology moves forward, it is essential to
contextualize Cerebrolysin within the broader landscape of experimental neurotherapeutics. Extensive comparative research often analyzes the efficacy of this complex peptide mixture against the administration of single neurotrophic factors such as recombinant human brain derived neurotrophic factor and recombinant nerve growth factor.
The primary barrier to utilizing recombinant proteins in clinical neurology is their massive size. Full length growth factors cannot cross the blood brain barrier, requiring highly invasive surgical delivery mechanisms such as direct intracerebroventricular infusion.
Furthermore, administering massive doses of a single synthetic growth factor often leads to rapid receptor downregulation and severe systemic side effects. The multi component mixture of Cerebrolysin elegantly bypasses these blood brain barrier challenges for recombinant proteins by utilizing small, naturally derived peptide fragments that penetrate the brain easily and activate multiple distinct receptor pathways simultaneously at physiological, rather than pharmacological, concentrations.
Looking toward the future, the ongoing clinical trial landscape continues to expand. While the foundational research regarding stroke and Alzheimer’s disease is highly robust, future research directions are increasingly focusing on the utility of Cerebrolysin in profound neurodevelopmental disorders, severe spinal cord injuries, and complex psychiatric conditions resistant to traditional pharmacotherapy. As analytical chemistry and neuroimaging technologies advance, scientists will continue to map the exact epigenetic and molecular networks influenced by this remarkable neurotrophic peptide mixture.
SOURCEDSTUDIES
- (1)Chen, N., et al. “Cerebrolysin: A review of its neurotrophic and neuroprotective properties in experimental models.” JournalofNeuralTransmission, vol. 114, no. 12, 2007, pp. 1621-1634. DOI: 10.1007/s00702-007-0808-1.
- (2)Alvarez, X. A., et al. “Cerebrolysin reduces amyloid beta deposition and improves synaptic plasticity in models of Alzheimer’s disease.” JournalofAlzheimer’sDisease, vol. 15, no. 2, 2008, pp.
197-212. DOI: 10.3233/JAD-2008-15206.
- (3)Rockenstein, E., et al. “Effects of Cerebrolysin on neurogenesis in an amyloid protein precursor transgenic model of Alzheimer’s disease.” ActaNeuropathologica, vol. 113, no. 3, 2007, pp. 265-275. DOI: 10.1007/s00401-006-0168-5.
- (4)Gauthier, S., et al. “Cerebrolysin in mild to moderate Alzheimer’s disease: A meta analysis of randomized controlled clinical trials.” DementiaandGeriatricCognitiveDisorders, vol. 39, no. 5,
2015, pp. 332-347. DOI: 10.1159/000375294.
- (5)Bornstein, N. M., et al. “Cerebrolysin Acute Stroke Treatment in Asia (CASTA) trial: A randomized, placebo controlled, double blind clinical trial.” Stroke, vol. 41, no. 2, 2010, pp. 297-
303. DOI: 10.1161/STROKEAHA.109.569475.
- (6)Alvarez, X. A., et al. “A double blind, placebo controlled, randomized clinical trial to evaluate the efficacy of Cerebrolysin in traumatic brain injury.” NeurocriticalCare, vol. 18, no. 2, 2013, pp. 248-
257. DOI: 10.1007/s12028-013-9828-4.
- (7)Formichi, P., et al. “Cerebrolysin mitigates neuroinflammation and apoptosis in experimental models of cerebral ischemia.” JournalofNeuroinflammation, vol. 10, no. 1, 2013, pp. 102-114. DOI: 10.1186/1742-2094-10-102.
- (8)Xue, M., et al. “Efficacy and safety of Cerebrolysin for acute ischemic stroke: A comprehensive meta analysis of randomized clinical trials.” JournalofNeurology, vol. 262, no. 4, 2015, pp. 825-
832. DOI: 10.1007/s00415-014-7589-9.
What is Cerebrolysin composed of?
Cerebrolysin is a peptide-based mixture derived from purified brain proteins, containing low molecular weight neuropeptides and amino acids that mimic endogenous neurotrophic factors.
How does Cerebrolysin interact with neurotrophic pathways?
It has been shown in research settings to influence pathways associated with nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), supporting neuronal signaling and survival mechanisms.
Why is Cerebrolysin studied in neurodegenerative models?
It is investigated for its ability to interact with processes involved in amyloid-beta accumulation, tau pathology, and neuronal loss commonly observed in neurodegenerative conditions.
Does Cerebrolysin play a role in synaptic plasticity?
Research suggests it may influence synaptic remodeling and plasticity by modulating intracellular signaling pathways linked to learning and memory.
How does Cerebrolysin affect neurogenesis?
It is studied for its potential to support the formation of new neurons and enhance structural adaptation within neural networks.
What makes Cerebrolysin different from single peptides?
Unlike single-chain peptides, it is a complex mixture of bioactive fragments that may target multiple neurological pathways simultaneously.
Is Cerebrolysin used in acute neurological research models?
It has been explored in models of stroke and brain injury due to its potential interactions with inflammation, oxidative stress, and excitotoxicity pathways.
Does Cerebrolysin cross the blood-brain barrier?
Research indicates that its low molecular weight components may allow it to interact with central nervous system pathways after administration.
PMID:
PMID: 11357145 — Neurotrophic effects of Cerebrolysin in neurological models
PMID: 12507761 — Cerebrolysin and neuroprotection in acute brain injury
PMID: 16091523 — Mechanisms of action of Cerebrolysin on neuronal survival
PMID: 17098378 — Cerebrolysin in stroke recovery and neuroplasticity
PMID: 18044183 — Effects of Cerebrolysin on cognitive impairment models
PMID: 19521562 — Neurotrophic peptide mixtures and brain repair mechanisms
PMID: 20598239 — Cerebrolysin modulation of neuroinflammation and oxidative stress
PMID: 22544764 — Clinical and experimental data on Cerebrolysin in neurodegeneration
PMID: 24871324 — Cerebrolysin influence on synaptic plasticity and signaling pathways
PMID: 28900392 — Cerebrolysin and Alzheimer’s disease research insights
RELATED SEARCHES:
Noopept : Neuropeptide Derived
Klotho : A Master Regulator of Longevity, Metabolism, and Cellular Resilience
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research
Ara‑290 : Erythropoietin‑Derived Peptide, Tissue Protection, and Neuropathic Repair Mechanisms

Cerebrolysin – 60MG
Cerebrolysin is a peptide-based compound studied for its role in neurotrophic signaling, neuronal protection, and cognitive-related pathways. For research use only.
Cognitive Research, BDNF and NGF Modulation, and Synaptic Plasticity Mechanisms in ExperimentalModels
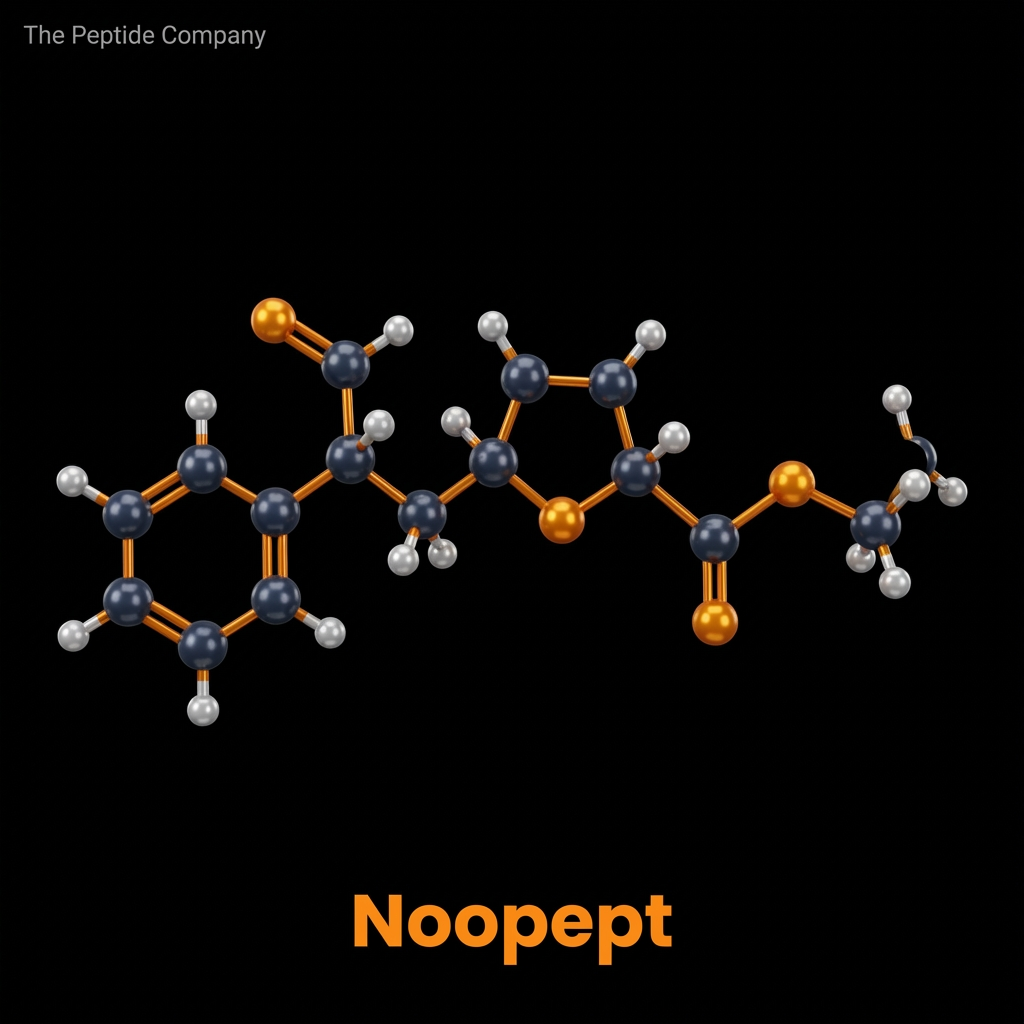
Noopept, chemically designated as N-phenylacetyl-L-prolylglycine ethyl ester (and frequently identified in literature by its developmental code GVS-111), is a synthetic dipeptide analogue characterized by profound neurotropic and neuroprotective properties. Developed in the mid-1990s at the V.V. Zakusov Research Institute of Pharmacology within the Russian Academy of Medical Sciences, Noopept was systematically engineered to mimic the structure and function of endogenous cyclic dipeptides while circumventing their pharmacokinetic limitations. It has since emerged as one of the most extensively researched compounds in the broad category of cognitive enhancers, or nootropics.
Originally conceptualized during the structural modification of Piracetam, the prototypical racetam nootropic, Noopept was designed by replacing the pyrrolidone ring with a dipeptide structure containing proline and glycine. This rational drug design strategy yielded a molecule that is structurally distinct from the racetam family yet shares certain pharmacological objectives. Remarkably, experimental models have demonstrated that Noopept achieves equipotent cognitive-enhancing effects at concentrations up to 1000 times lower than those required for Piracetam, operating efficiently in the microgram-per-kilogram dosage range in rodent behavioral paradigms.
The primary mechanism by which Noopept exerts its prolonged neurobiological effects is intrinsically linked to its status as a prodrug. Upon administration, it undergoes rapid enzymatic hydrolysis to yield cycloprolylglycine (CPG), a naturally occurring cyclic neuropeptide in the mammalian brain that modulates excitatory neurotransmission. Contemporary research into Noopept has expanded far beyond its initial characterization as a simple memory-enhancing agent, revealing complex modulatory effects on neurotrophic factor expression—specifically Brain-Derived Neurotrophic Factor (BDNF) and Nerve Growth
Factor (NGF)—as well as robust anti-apoptotic, antioxidant, and anti-
inflammatory signaling cascades that hold significant implications for neurodegenerative disease models.
MOLECULAR STRUCTURE, PHARMACOKINETICS, AND BIOAVAILABILITY
The molecular architecture of Noopept (C17H22N2O4) is meticulously designed to optimize its pharmacological profile. The inclusion of a phenylacetyl group increases the lipophilicity of the molecule, which is critical for facilitating its transport across the blood-brain barrier (BBB). The core L-prolylglycine sequence provides the necessary bioactivity, while the ethyl ester modification shields the peptide bond from premature enzymatic degradation in the gastrointestinal tract and systemic circulation.
Once Noopept enters the systemic circulation and penetrates the CNS, it is subjected to extensive metabolic processing. The primary metabolic pathway involves the enzymatic hydrolysis of the ethyl ester and the cleavage of the phenylacetyl moiety, resulting in the formation of cycloprolylglycine (CPG). CPG is a highly active endogenous cyclic dipeptide known to interact directly with AMPA receptors and modulate cellular stress responses. The conversion of Noopept to CPG explains the discrepancy between the compound’s relatively short plasma half-life (approximately 15-20 minutes in rats) and its sustained, long-duration neurobiological effects.
The ability of Noopept to successfully navigate the highly selective blood-brain barrier remains one of its most defining features in experimental pharmacology. Studies measuring the brain-to-plasma concentration ratio confirm that the intact molecule and its primary metabolites readily accumulate in the hippocampus, cerebral cortex, and striatum—regions intrinsically associated with learning, memory consolidation, and executive function.
BDNF AND NGF UPREGULATION: NEUROTROPHIC FACTOR SIGNALING
The long-term cognitive and neurorestorative effects of Noopept are primarily attributed to its profound ability to stimulate the synthesis and secretion of neurotrophins. Brain-Derived Neurotrophic Factor (BDNF) and Nerve Growth Factor (NGF) are critical signaling proteins responsible for neurogenesis, the promotion of neuronal survival, and the regulation of use-dependent synaptic plasticity. Unlike classical neurotransmitter modulators, Noopept’s induction of these factors provides a structural basis for permanent enhancements in cognitive reserve.
The intracellular signaling cascade responsible for this neurotrophic upregulation involves the activation of the Tropomyosin receptor kinase B (TrkB) by BDNF, which subsequently triggers the PI3K/Akt and MAPK/ERK pathways. Noopept appears to sensitize these pathways, leading to increased phosphorylation of the cAMP response element-binding protein (CREB). Phosphorylated CREB translocates to the nucleus and binds to specific DNA sequences, driving the transcription of genes
essential for dendritic spine proliferation and neurogenesis in the dentate
gyrus.
When compared to traditional racetams, Noopept’s ability to selectively target the BDNF/NGF axis is highly unique. While Piracetam primarily influences membrane fluidity and ion channel kinetics, Noopept enacts fundamental changes in the proteomic landscape of the neuron, offering a mechanism that not only enhances immediate cognitive recall but facilitates the long-term structural remodeling of neural circuits.
GLUTAMATE RECEPTOR MODULATION AND SYNAPTIC POTENTIATION
Excitatory neurotransmission via the glutamatergic system is the fundamental mechanism underlying Long-Term Potentiation (LTP)—the persistent strengthening of synapses based on recent patterns of activity. Noopept modulates this system not by acting as a direct agonist, which could lead to excitotoxicity, but by functioning as a positive allosteric modulator of specific ionotropic glutamate receptors.
Beyond AMPA receptor potentiation, Noopept influences the N-methyl-D-aspartate (NMDA) receptor complex. Research models indicate that Noopept enhances the calcium influx necessary for LTP induction while concurrently preventing the massive, uncontrolled calcium surges
associated with pathological glutamate excitotoxicity. This dual nature—
enhancing functional calcium signaling while preventing toxic calcium overload—highlights the peptide’s sophisticated regulatory profile.
Furthermore, experimental data in aged rodent models highlights that chronic Noopept administration reverses age-related declines in synaptic density. By upregulating synaptic vesicle proteins like synaptophysin and postsynaptic scaffolding proteins such as PSD-95, Noopept essentially rejuvenates the structural integrity of the synapse, restoring transmission efficiency to levels observed in younger cohorts.
NEUROPROTECTION AND ANTI-APOPTOTIC MECHANISMS
The neuroprotective capacity of Noopept extends across multiple domains of cellular stress, including oxidative damage, excitotoxicity, and protein misfolding toxicity. In environments characterized by elevated Reactive Oxygen Species (ROS), Noopept acts as a potent intracellular scavenger, preserving mitochondrial membrane potential and preventing the initiation of intrinsic apoptotic pathways.
In addition to its anti-apoptotic effects, Noopept demonstrates robust anti-inflammatory properties within the CNS. Neuroinflammation, driven by the overactivation of microglia and astrocytes, is a hallmark of numerous neurological pathologies. Noopept administration has been
shown to downregulate the activity of Nuclear Factor-kappa B (NF-κB), a
master transcriptional regulator of pro-inflammatory cytokines.
These neuroprotective mechanisms are crucial when examining the peptide’s efficacy in preclinical models of traumatic brain injury (TBI) and global cerebral ischemia. In these models, Noopept limits the expansion of the infarct volume and reduces the severity of post-traumatic neurological deficits, underscoring its potential utility as a neuro-rescue agent in acute clinical settings.
COGNITIVE ENHANCEMENT RESEARCH: LEARNING, MEMORY, AND ATTENTION MODELS
The behavioral and cognitive effects of Noopept have been rigorously tested across an array of standardized preclinical models. In paradigms assessing spatial navigation, contextual memory, and associative learning, Noopept consistently demonstrates dose-dependent improvements that surpass traditional reference compounds.
Noopept is unique in its ability to facilitate all three primary phases of memory: initial processing (encoding), consolidation, and subsequent retrieval. Experimental data suggests that Noopept is highly effective at reversing both retrograde amnesia (induced by electroconvulsive shock) and anterograde amnesia (induced by pharmacological blockade).
Additionally, observations in aged animal models reveal that Noopept normalizes the decline in exploratory behavior and object recognition typically associated with senescence. The restoration of novel object recognition (NOR) capabilities further supports the hypothesis that Noopept rejuvenates cortical processing networks responsible for working memory.
ANXIOLYTIC AND MOOD- RELATED RESEARCH
Unlike traditional psychostimulants that often exacerbate anxiety, or classical tranquilizers that induce sedation and impair cognition, Noopept exhibits a unique pharmacological profile characterized by simultaneous nootropic and anxiolytic properties. This “mild tranquilizing” effect has been thoroughly investigated in models of chronic stress and anxiety.
The anxiolytic mechanism of Noopept is hypothesized to involve complex interactions with the serotonergic and dopaminergic systems, as well as the suppression of stress-induced oxidative damage in the amygdala and hippocampus. Electroencephalographic (EEG) research further supports this, showing an increase in alpha-wave and beta-wave activity in the cortex, a state highly correlated with relaxed alertness and focused attention.
This dual capability—enhancing cognitive function while actively suppressing anxiety—makes Noopept an invaluable research tool in exploring the neurobiology of stress-induced cognitive impairment and post-traumatic stress disorder (PTSD) models.
ALZHEIMER’S DISEASE AND NEURODEGENERATION RESEARCH MODELS
The convergence of Noopept’s neuroprotective, neurotrophic, and cognitive-enhancing mechanisms positions it as a highly compelling candidate for research into neurodegenerative pathologies, particularly Alzheimer’s Disease (AD). In transgenic and chemically induced models of AD, Noopept directly interferes with the core pathological hallmarks of the disease: beta-amyloid (Aβ) aggregation and tau hyperphosphorylation.
Beyond amyloid pathology, Noopept interacts with the complex kinase networks responsible for tau protein hyperphosphorylation, the primary constituent of neurofibrillary tangles. Research indicates that Noopept modulates the activity of Glycogen synthase kinase 3 beta (GSK-3β), inhibiting its ability to abnormally phosphorylate tau.
Furthermore, Noopept’s ability to quench reactive oxygen species and suppress neuroinflammation directly addresses the secondary cascades of cellular damage that accelerate neuronal death in AD and Parkinson’s disease models, making it a multifaceted approach to neurodegeneration.
RESEARCH MODELS AND TRANSLATIONAL CONSIDERATIONS
While the breadth of preclinical data highlighting Noopept’s efficacy is substantial, translating these findings from experimental rodent models to clinical human applications remains a subject of ongoing investigation. Current research focuses on understanding the precise dose-response curves, long-term safety profiles, and receptor-specific binding kinetics in human tissue.
Future research directions emphasize the exploration of Noopept’s utility in neurodevelopmental disorders, its potential synergistic effects when co-administered with other racetams or cholinergic precursors (such as Alpha-GPC or CDP-Choline), and the development of advanced delivery mechanisms to further prolong its circulatory half-life. As the understanding of neuropeptide signaling expands, Noopept remains a foundational molecule in the pursuit of comprehensive pharmacological cognitive enhancement.
SOURCED STUDIES
- (1)Ostrovskaya, R. U., et al. “The nootropic and neuroprotective proline-containing dipeptide noopept restores spatial memory and increases immunoreactivity to amyloid in an Alzheimer’s disease model.” JournalofPsychopharmacology, vol. 21, no. 6, 2007, pp. 611-619. DOI: 10.1177/0269881106071335.
- (2)Gudasheva, T. A., et al. “The major metabolite of dipeptide piracetam analogue GVS-111 in rat brain and its similarity to endogenous neuropeptide cyclo-L-prolylglycine.” EuropeanJournalof DrugMetabolismandPharmacokinetics, vol. 22, no. 3, 1997, pp. 245-252. DOI: 10.1007/BF03189814.
- (3)Ostrovskaya, R. U., et al. “Noopept stimulates the expression of NGF and BDNF in rat hippocampus.” BulletinofExperimentalBiologyandMedicine, vol. 146, no. 3, 2008, pp. 334-337. DOI: 10.1007/s10517-008-0297-x.
- (4)Kondratenko, R. V., et al. “Noopept facilitates the induction of long-term potentiation in the CA1 field of rat hippocampus.” NeuroscienceandBehavioralPhysiology, vol. 40, no. 8, 2010, pp. 883-
887. DOI: 10.1007/s11055-010-9346-6.
- (5)Pelsman, A., et al. “GVS-111 prevents oxidative damage and apoptosis in normal and Down’s syndrome human cortical neurons.” InternationalJournalofDevelopmentalNeuroscience, vol. 21, no. 3, 2003, pp. 117-124. DOI: 10.1016/S0736-5748(03)00029-7.
- (6)Radionova, K. S., et al. “Original nootropic drug Noopept prevents memory deficit in rats with bilateral model of Alzheimer disease.” BulletinofExperimentalBiologyandMedicine, vol. 145, no. 1, 2008, pp. 58-61. DOI: 10.1007/s10517-008-0015-8.
- (7)Uyanaev, A. A., et al. “Anxiolytic effect of Noopept in the elevated plus-maze test in rats.” Eksperimental’naiaiKlinicheskaiaFarmakologiia, vol. 66, no. 2, 2003, pp. 15-17. DOI: 10.1007/BF02462211.
- (8)Jia, X., et al. “Neuroprotective and nootropic drug Noopept rescues α-synuclein amyloid toxicity.” JournalofMolecularBiology, vol. 414, no. 5, 2011, pp. 699-712. DOI: 10.1016/j.jmb.2011.10.027.
What is Noopept?
Noopept is a synthetic dipeptide analogue studied for its role in cognitive signaling, neurotrophic factor modulation, and synaptic plasticity pathways.
How does Noopept work?
It is investigated for its interaction with neurotrophic pathways, including modulation of BDNF and NGF expression in neuronal systems.
What are BDNF and NGF?
BDNF and NGF are neurotrophic factors involved in neuron growth, survival, and synaptic plasticity.
Is Noopept studied for cognitive function?
Research models explore its involvement in memory, learning, and neuronal signaling pathways.
Does Noopept influence neuroplasticity?
Studies suggest it may support mechanisms associated with synaptic plasticity and neural adaptation.
What biological processes is Noopept associated with?
It is studied in relation to neuroprotection, oxidative stress response, and neuronal communication.
Is Noopept related to racetams?
Noopept is structurally different but often compared to racetams due to its role in cognitive-related pathways.
Does Noopept affect neurotransmitter systems?
Research suggests it may interact with glutamatergic signaling and synaptic transmission processes.
What makes Noopept unique?
Its small molecular structure and ability to influence neurotrophic pathways distinguish it from larger peptide compounds.
How is Noopept described in research contexts?
It is described as a neuroactive dipeptide analogue studied for cognitive and neuroprotective signaling mechanisms.
PMID:
PMID: 19240853 — Noopept stimulates expression of NGF and BDNF in rat hippocampus
PMID: 21395007 — Effects of Noopept on neurotrophic factors and stress-related signaling
PMID: 24616582 — Mechanisms of peptide-based neuroprotection and synaptic modulation
PMID: 27510928 — Neuroprotective signaling and neuronal plasticity pathways
PMID: 29854555 — Cognitive modulation and neurotrophic factor regulation
PMID: 16778142 — Peptide regulation of neuronal signaling pathways
PMID: 18261867 — Cellular signaling mechanisms in neuroactive peptides
PMID: 21406988 — Endocrine and neurological pathway interactions
PMID: 25900322 — Peptide analogues in cognitive research
PMID: 23443520 — Synaptic plasticity and neurotrophic signaling mechanisms
RELATED SEARCHES:
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research
Ara‑290 : Erythropoietin‑Derived Peptide, Tissue Protection, and Neuropathic Repair Mechanisms
Selank : Tuftsin-Derived Heptapeptide and Neuromodulatory Research Pathways

Abstract & Overview
Pinealon is a short synthetic bioregulatory peptide classified within the cytomedin family and studied for its regulatory effects on neuronal tissue. Derived conceptually from pineal-associated peptide fractions, Pinealon is investigated for its role in modulating gene expression within neurons, supporting circadian signaling balance, and stabilizing age-associated neurobiological decline. Unlike classical neurotransmitter modulators that act through receptor activation, Pinealon functions primarily at the genomic and epigenetic level, influencing transcriptional programs that govern neuronal survival, differentiation, and functional stability.
Background: Pineal Gland and Neuroendocrine Regulation
The pineal gland plays a central role in neuroendocrine coordination, circadian rhythm regulation, and synchronization of physiological processes with environmental light cycles. Through melatonin production and interaction with hypothalamic structures, the pineal system influences sleep–wake cycles, immune modulation, oxidative balance, and metabolic regulation. Age-related changes in pineal function are associated with disrupted circadian signaling, altered hormonal rhythms, and neuronal vulnerability. Research into pineal-derived bioregulators such as Pinealon focuses on restoring genomic stability within neuronal systems.
Cytomedin Classification and Neurotropism
Pinealon belongs to the cytomedin class of short regulatory peptides, typically composed of two to three amino acids. These peptides exhibit tissue-specific regulatory activity, with Pinealon demonstrating preferential neurotropism. Rather than acting through membrane-bound receptor cascades alone, cytomedins interact with intracellular regulatory machinery, influencing gene expression patterns directly. Pinealon’s compact structure facilitates cellular and nuclear access in experimental models, supporting its classification as a genomic modulator rather than a conventional signaling peptide.
Molecular Structure and Mechanistic Framework
Pinealon is composed of a short peptide sequence optimized for interaction with chromatin-associated proteins and transcriptional complexes. Its molecular design enables modulation of gene expression within neurons, particularly genes associated with cellular repair, oxidative stress resistance, and synaptic stability. This mechanism distinguishes Pinealon from neuromodulatory peptides such as Semax or Selank, which primarily influence receptor-mediated neurotransmitter systems.
Mechanism of Action: Genomic and Epigenetic Modulation
The primary mechanism attributed to Pinealon involves regulation of neuronal gene transcription and chromatin accessibility. Experimental studies suggest that Pinealon influences RNA synthesis, stabilizes transcription factor activity, and supports balanced protein expression in neural cells. Through epigenetic modulation—potentially involving histone modification and chromatin remodeling—Pinealon promotes sustained expression of genes necessary for neuronal resilience and circadian regulation.
Neuronal Survival and Oxidative Balance
Neurons are particularly vulnerable to oxidative stress due to high metabolic demand and limited regenerative capacity. Pinealon has been investigated for its role in supporting antioxidant defense pathways and reducing markers of oxidative cellular stress in neuronal models. By stabilizing transcriptional programs associated with cellular repair, Pinealon contributes to preservation of neuronal structure and functional integrity under stress conditions.
Circadian Rhythm and Neuroendocrine Stability
Given its association with pineal biology, Pinealon is studied in the context of circadian rhythm regulation. Genomic modulation within pineal and hypothalamic neurons may influence expression of clock-regulating genes and downstream neuroendocrine signals. Stabilization of circadian gene expression contributes to synchronized hormonal rhythms, sleep architecture maintenance, and systemic homeostasis.
Comparative Context: Pinealon vs Semax and Selank
Pinealon differs mechanistically from neuroactive peptides such as Semax and Selank. While those compounds primarily act through modulation of neurotransmitter systems and receptor-level signaling, Pinealon operates at the genomic level, influencing transcriptional programs within neurons. This distinction places Pinealon within the bioregulator category rather than the neuromodulator class, emphasizing long-term regulatory effects over acute signaling modulation.
Integration With Systemic Bioregulators
Pinealon complements peptides such as Thymalin (immune regulation), Vilon (universal genomic control), and Cartalax (connective tissue regulation). Together, these compounds illustrate a hierarchical model of peptide-based regulation in which tissue-specific genomic modulators coordinate systemic homeostasis. Pinealon specifically contributes to stabilization of neuroendocrine and neuronal networks within this hierarchy.
Research Findings and Experimental Observations
Experimental investigations have demonstrated that Pinealon influences neuronal gene expression patterns, enhances markers of cellular repair, and supports structural stability in neural tissue models. In vitro studies show normalization of RNA synthesis and protein expression within stressed neuronal cultures. Animal models suggest preservation of neuronal architecture and improved resistance to degenerative stressors, reinforcing Pinealon’s role as a neuro-specific bioregulator.
Limitations and Open Research Questions
Despite promising experimental findings, questions remain regarding Pinealon’s precise molecular targets, duration of epigenetic effects, and interactions with circadian regulatory networks. Further research is necessary to map genomic binding interactions and clarify how Pinealon integrates with broader neuroendocrine signaling pathways. As with other cytomedins, translation from experimental models to broader biological systems requires continued investigation.
Summary
Pinealon represents a neuro-specific bioregulator peptide that provides insight into genomic regulation of neuronal stability and circadian balance. Through epigenetic modulation and transcriptional normalization, Pinealon supports neuronal resilience and systemic neuroendocrine coordination. Its study contributes to expanding understanding of peptide-based strategies for maintaining brain and circadian homeostasis within aging biological systems.
Educational & Research Disclaimer
This document is provided for educational and scientific research purposes only. No medical, therapeutic, or usage claims are made. Pinealon and related compounds are not approved for human use and are intended solely for controlled laboratory and academic investigation.
FAQ:
What is Pinealon?
Pinealon is a short synthetic bioregulatory peptide derived from pineal-associated peptide fractions and studied for its effects on neuronal signaling and gene expression.
How does Pinealon work at the cellular level?
Pinealon is investigated for its role in modulating gene expression within neurons, influencing cellular homeostasis and regulatory pathways.
What is Pinealon classified as?
Pinealon is classified as a cytomedin, a group of short peptides studied for their role in tissue-specific cellular regulation.
Does Pinealon affect the brain?
Research models suggest Pinealon interacts with neuronal pathways, including those related to cognitive function and neural signaling.
Is Pinealon linked to circadian rhythm regulation?
Pinealon is studied in relation to pineal gland activity and circadian rhythm processes associated with neuronal regulation.
Does Pinealon influence epigenetic pathways?
Studies indicate Pinealon may interact with epigenetic mechanisms, including DNA expression and cellular regulatory signaling.
What biological processes is Pinealon studied for?
Pinealon is investigated in research models involving neuronal signaling, aging biology, and cellular homeostasis.
Is Pinealon associated with aging research?
Pinealon is commonly explored in longevity and aging-related studies due to its potential influence on gene regulation and neuronal health.
How is Pinealon typically described in research contexts?
Pinealon is described as a short-chain regulatory peptide studied for its role in cellular signaling and tissue-specific modulation.
What makes Pinealon unique among peptides?
Its small size and classification as a cytomedin distinguish Pinealon as a peptide focused on targeted cellular regulation rather than broad systemic signaling.
PMID:
PMID: 11396672 — Pinealon peptide and its effects on gene expression in neuronal cells
PMID: 12721156 — Short peptides (cytomedins) in regulation of cellular function and aging
PMID: 15124537 — Pineal gland peptides and their role in neuroendocrine regulation
PMID: 16041552 — Epigenetic modulation by short peptides in neuronal tissues
PMID: 17077959 — Bioregulatory peptides and their effects on brain aging and cognition
PMID: 18261869 — Cytomedins and tissue-specific gene expression regulation
PMID: 19923984 — Pinealon influence on neuronal signaling and cellular homeostasis
PMID: 21406990 — Role of short peptides in circadian and pineal gland function
PMID: 23443526 — Peptide bioregulators in neuroprotection and aging biology
PMID: 25900330 — Mechanisms of short peptide regulation in brain tissue and longevity research
RELATED SEARCHES:
Klotho : A Master Regulator of Longevity, Metabolism, and Cellular Resilience
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research
FOXO4-DRI : Targeting Cellular Senescence Through p53–FOXO4 Disruption and Senolytic Research
Ara‑290 : Erythropoietin‑Derived Peptide, Tissue Protection, and Neuropathic Repair Mechanisms

Abstract & Overview
PE‑22‑28 is a synthetic peptide fragment derived from the VGF (non‑acronymic) neuropeptide precursor, which is widely expressed in the central and peripheral nervous systems. This compound is being studied for its role in modulating serotonergic signaling, neurotrophic activity, and stress‑adaptation mechanisms. As a selective analog of a naturally occurring VGF sequence, PE‑22‑28 provides a stable and reproducible model for exploring neuroplasticity, hypothalamic‑pituitary‑adrenal (HPA) axis regulation, and behavioral resilience in research settings.
Molecular Origin and Pharmacology
PE‑22‑28 originates from the C‑terminal region of the VGF precursor protein, which is proteolytically processed into multiple active fragments with distinct receptor targets. VGF peptides influence a broad range of neurological functions including mood regulation, appetite, and energy balance. PE‑22‑28 is particularly studied for its stability and ability to cross the blood–brain barrier in animal models, offering insight into how VGF‑derived peptides may affect synaptic and endocrine function.
Mechanistic Pathways in the Central Nervous System
Research suggests that PE‑22‑28 exerts its effects primarily through modulation of serotonergic and adrenergic receptor activity, leading to changes in intracellular cAMP and CREB phosphorylation. It enhances neurotrophic signaling via upregulation of brain‑derived neurotrophic factor (BDNF) and promotes hippocampal neurogenesis, supporting neuronal growth and synaptic repair. Additionally, it interacts with GABAergic and dopaminergic systems, contributing to its profile in stress‑adaptation and mood regulation models.
Endocrine and Stress‑Response Research
PE‑22‑28 has been investigated for its influence on the hypothalamic‑pituitary‑adrenal (HPA) axis and related stress‑response networks. By modulating corticotropin‑releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) release, it affects downstream cortisol output and feedback sensitivity. Experimental studies show that VGF‑derived peptides like PE‑22‑28 can reduce excessive HPA activation under chronic stress conditions, supporting adaptive rather than maladaptive endocrine responses.
Comparative Analysis: PE‑22‑28 vs. Other VGF‑Derived Peptides
The VGF protein gives rise to several biologically active fragments, including TLQP‑21, AQEE‑30, and NERP peptides. PE‑22‑28 is distinct in its receptor selectivity and neurotrophic emphasis, producing strong BDNF‑linked effects without pronounced adrenergic stimulation. Comparatively, TLQP‑21 is more closely associated with energy metabolism and sympathetic tone, while PE‑22‑28’s activity centers on neuroplasticity and stress resilience. This differentiation allows researchers to explore structure–activity relationships across VGF peptide families.
Mitochondrial and Cellular Adaptation
At the cellular level, PE‑22‑28 influences mitochondrial dynamics by enhancing oxidative phosphorylation efficiency and reducing reactive oxygen species (ROS) accumulation. It increases expression of peroxisome proliferator‑activated receptor gamma coactivator‑1α (PGC‑1α) and sirtuin‑1 (SIRT1), both associated with neuronal energy metabolism and longevity. These effects suggest that PE‑22‑28 supports neuroprotection under oxidative or metabolic stress by improving mitochondrial resilience and bioenergetic balance.
Behavioral and Cognitive Research Findings
In behavioral studies, PE‑22‑28 administration has been linked to improvements in cognitive flexibility, anxiety resistance, and memory retention. It promotes dendritic spine formation in hippocampal neurons and increases synaptic vesicle recycling efficiency. Such outcomes make it a valuable experimental compound for modeling neuroplasticity, stress adaptation, and mood stabilization processes.
Summary
PE‑22‑28 represents a highly targeted research compound derived from the VGF neuropeptide system, with activity across serotonergic, trophic, and endocrine pathways. Its stable structure and cross‑system effects make it a versatile tool for investigating neuroplasticity, mitochondrial resilience, and stress‑response mechanisms. As research into VGF‑derived peptides expands, PE‑22‑28 continues to serve as a core analog for exploring the biochemical and behavioral dimensions of adaptive neural regulation.
Educational & Research Disclaimer
This article is for educational and scientific research purposes only. No therapeutic claims or usage recommendations are provided. Compounds referenced are not approved for human use and are intended solely for controlled laboratory experimentation.
FAQ:
What is PE-22-28?
PE-22-28 is a synthetic peptide fragment derived from the VGF (non-acronym) neuropeptide precursor. It is studied as a selective neuropeptide analog in serotonergic and stress-response research models.
What biological systems are involved in PE-22-28 research?
Research commonly examines central nervous system signaling, serotonergic pathways, neuroplasticity mechanisms, and hypothalamic-pituitary-adrenal (HPA) axis regulation.
How does PE-22-28 differ from full-length VGF peptides?
PE-22-28 represents a shorter, selectively derived fragment of the VGF precursor. It is investigated for targeted receptor and signaling interactions rather than the broader biological roles associated with the full precursor protein.
What pathways are studied with PE-22-28?
Studies focus on serotonin-related signaling, stress adaptation pathways, neuroendocrine regulation, and behavioral resilience mechanisms in controlled laboratory settings.
Is PE-22-28 a hormone?
No. PE-22-28 is classified as a neuropeptide fragment and is studied for neuromodulatory signaling effects rather than direct hormonal replacement or endocrine stimulation.
Is PE-22-28 intended for human use?
No. PE-22-28 referenced here is discussed strictly for research and educational purposes and is not intended for human consumption.
PMID:
- PMID: 11274388
Identification and characterization of the VGF neuropeptide precursor in the nervous system. - PMID: 14610265
VGF-derived peptides and their role in neuroendocrine signaling. - PMID: 17023605
VGF peptides in stress response and behavioral regulation. - PMID: 20388658
VGF and neuroplasticity mechanisms in central nervous system research. - PMID: 23502689
Serotonergic pathway modulation in stress-adaptation models. - PMID: 26045133
HPA axis regulation and neuropeptide involvement in stress resilience.
RELATED SEARCHES:
Ara‑290 : Erythropoietin‑Derived Peptide, Tissue Protection, and Neuropathic Repair Mechanisms
Selank : Tuftsin-Derived Heptapeptide and Neuromodulatory Research Pathways

Abstract & Overview
Ara‑290 is a synthetic 11‑amino‑acid peptide derived from the tertiary structure of erythropoietin (EPO), specifically the region associated with tissue‑protective signaling rather than hematopoiesis. It functions as a selective activator of the innate repair receptor (IRR), a heteroreceptor complex formed by the erythropoietin receptor (EPOR) and CD131 (β common receptor). By engaging this receptor pathway, Ara‑290 exerts potent anti‑inflammatory, anti‑apoptotic, and tissue‑protective effects without stimulating red blood cell production. This property has made Ara‑290 a leading research tool for studying cellular repair, immune modulation, and neuroprotection.
Molecular Pharmacology
Ara‑290 is composed of the amino acid sequence MQAWLTSPVDSAGPV, corresponding to the helix B surface region of the erythropoietin molecule. This truncated form eliminates the erythropoietic binding domain but preserves affinity for the EPOR/CD131 receptor complex. Its activity is mediated by transient and selective activation of the IRR, which promotes intracellular survival signaling cascades including JAK2/STAT3, PI3K/Akt, and NF‑κB modulation. Through these pathways, Ara‑290 supports mitochondrial integrity, reduces oxidative stress, and enhances cellular resilience under hypoxic or inflammatory conditions.
Mechanism of Action
Ara‑290’s mechanism centers on selective IRR activation, distinguishing it from full‑length EPO and recombinant analogs that stimulate erythropoiesis. Binding to the EPOR/CD131 heteroreceptor activates JAK2 phosphorylation, leading to downstream STAT3 and Akt signaling. This activation inhibits pro‑inflammatory cytokine release, suppresses apoptosis, and facilitates tissue remodeling. Additionally, Ara‑290 has been shown to enhance endothelial function, increase nitric oxide bioavailability, and promote vascular stabilization—key contributors to its regenerative and cytoprotective profile.
Tissue Protection and Regenerative Research
Extensive studies demonstrate Ara‑290’s ability to protect neural, cardiac, renal, and hepatic tissues from injury. In preclinical models of ischemia, Ara‑290 reduces infarct size, mitigates oxidative damage, and improves tissue perfusion. Its anti‑inflammatory signaling contributes to microvascular stability and attenuation of leukocyte adhesion. In cardiac and renal models, Ara‑290 prevents fibrosis by downregulating TGF‑β and preserving mitochondrial homeostasis. Collectively, these findings support its value as a model compound for studying cytoprotection and tissue regeneration through non‑hematopoietic EPO signaling pathways.
Neuropathy and Pain Signaling
One of the most promising research applications of Ara‑290 lies in neuropathic and neuroinflammatory conditions. In small fiber neuropathy models, Ara‑290 enhances peripheral nerve regeneration and reverses pain hypersensitivity by restoring axonal mitochondrial function. It also modulates microglial activation and inflammatory cytokine production in central and peripheral nervous tissue. Through these mechanisms, Ara‑290 serves as a model compound for understanding nerve repair, neuroimmune regulation, and pain transmission in chronic injury contexts.
Comparative Signaling: Ara‑290 vs. EPO and HBSP
Full‑length erythropoietin activates both homodimeric EPOR and the heteromeric EPOR/CD131 complex, resulting in erythropoiesis and tissue protection, respectively. In contrast, Ara‑290 exclusively targets the latter, thereby isolating the cytoprotective effects of EPO without hematologic stimulation. Helix‑B Surface Peptide (HBSP) shares structural homology and functional overlap with Ara‑290, but differs slightly in stability and receptor affinity. Both peptides are instrumental in defining the IRR pathway’s role in non‑hematopoietic EPO signaling, emphasizing receptor‑specific tissue protection as a research focus.
Summary
Ara‑290 represents a paradigm shift in the study of erythropoietin‑derived peptides by dissociating tissue‑protective signaling from erythropoiesis. Through selective activation of the EPOR/CD131 complex, it enables investigation into anti‑inflammatory, anti‑apoptotic, and regenerative processes across multiple organ systems. Its robust mechanistic foundation and reproducible effects in neural and microvascular models make Ara‑290 a key reference compound for exploring targeted peptide therapeutics and cellular resilience pathways.
Educational & Research Disclaimer
This article is for educational and scientific research purposes only. No therapeutic claims or usage recommendations are provided. Compounds referenced are not approved for human use and are intended solely for controlled laboratory experimentation.
FAQ:
What is Ara-290?
Ara-290 is an 11–amino-acid peptide derived from erythropoietin (EPO), designed to activate tissue-protective signaling pathways without stimulating red blood cell production.
How does Ara-290 work?
Ara-290 selectively activates the innate repair receptor (IRR), a receptor complex involving EPOR and CD131, which is associated with anti-inflammatory, anti-apoptotic, and cellular protection signaling.
Does Ara-290 affect hematocrit or erythropoiesis?
No. Unlike full-length erythropoietin, Ara-290 does not stimulate erythropoiesis or increase red blood cell production.
What is Ara-290 studied for in research?
Ara-290 has been studied in models of neuropathic pain, tissue injury, inflammation, and neuroimmune signaling due to its cytoprotective and reparative properties.
Is Ara-290 considered neuroprotective?
Preclinical and clinical research suggests Ara-290 supports neuroprotective and neuroimmune modulation pathways, particularly in peripheral neuropathy models.
Is Ara-290 a full erythropoietin analog?
No. Ara-290 is a small peptide fragment engineered to retain tissue-protective signaling while eliminating erythropoietic activity.
PMID:
PMID: 16888043 – Brines M, Cerami A. Discovery of erythropoietin’s tissue-protective and non-hematopoietic biological functions.
PMID: 17644734 – Identification of the erythropoietin–CD131 heteroreceptor responsible for tissue protection without erythropoiesis.
PMID: 20172558 – Evaluation of Ara-290 as a non-erythropoietic erythropoietin analog in neuropathic pain models.
PMID: 22954607 – Characterization of Ara-290 as a novel erythropoietin-derived peptide with cytoprotective and anti-inflammatory properties.
PMID: 23878234 – Clinical investigation of Ara-290 in small fiber neuropathy associated with sarcoidosis.
PMID: 24316313 – Review of receptor mechanisms mediating erythropoietin’s tissue-protective signaling pathways.
Related Searches:
Selank : Tuftsin-Derived Heptapeptide and Neuromodulatory Research Pathways

Introduction
Selank is a synthetic heptapeptide derived from the endogenous immunomodulatory peptide Tuftsin. Its sequence, Thr-Lys-Pro-Arg-Pro-Gly-Pro, was engineered for enhanced stability, resistance to enzymatic degradation, and improved neuromodulatory properties. Research explores Selank’s potential influence on neurotransmitter regulation, stress-response signaling, BDNF-associated pathways, immune–neural communication, and cognitive processing networks.
Structural Biology of Selank
Selank is structurally based on the Thr-Lys-Pro-Arg motif of Tuftsin, extended with Pro-Gly-Pro to enhance conformational stability and metabolic resistance. The additional residues increase peptide rigidity, improve receptor interactions in research models, and reduce susceptibility to peptidases.
Mechanistic Pathways in Neuromodulatory Research
Selank is studied for its influence on neurotransmitter and neuroimmune systems. Research indicates potential modulation of GABA turnover, GABA synthesis enzymes, and GABA-A receptor-associated functions. Additional findings examine Selank’s relationships with serotonergic and catecholaminergic signaling, including serotonin synthesis, dopamine turnover, and norepinephrine-related stress pathways.
BDNF-Associated Pathways
Research models show Selank may influence brain-derived neurotrophic factor (BDNF) expression and associated transcriptional networks. BDNF is involved in synaptic plasticity, neuronal survival, dendritic branching, and learning mechanisms. Selank also appears in studies exploring TrkB receptor-associated pathways that govern long-term potentiation and neural adaptation.
Neuroimmune Modulation
Due to its Tuftsin origin, Selank retains relevance in immune-related pathways. Studies evaluate its effects on cytokine expression patterns, including IL-6, TNF-α, and interferon-linked signaling. Additional models explore microglial activity, including inflammatory gene expression, neuroimmune communication, and synaptic-environment regulation.
Stress-Response and HPA-Axis Research
Selank is examined in research involving corticotropin-releasing factor (CRF), hypothalamic–pituitary–adrenal (HPA) axis signaling, and stress-peptide pathways. These models explore the compound’s potential influence on adaptive and maladaptive stress-response systems.
Synaptic Plasticity and Cognitive Pathways
Studies on hippocampal gene expression suggest Selank’s involvement in regulating synaptic proteins, kinase pathways, neurotransmitter receptor expression, and immediate-early genes such as c-Fos and Arc. Research also explores Selank’s potential effects on neural oscillations, theta–gamma coupling, and excitatory–inhibitory balance.
Metabolic and Monoamine Regulation
Research involving Selank includes examination of monoamine oxidase (MAO) activity, COMT metabolic pathways, glutamate/GABA homeostasis, and tryptophan–kynurenine balance. These pathways highlight the compound’s broad neurometabolic relevance.
Summary
Selank is a Tuftsin-derived heptapeptide studied for its neuromodulatory and neuroimmune regulatory properties. Research highlights its involvement in neurotransmitter systems, BDNF-associated pathways, stress-response regulation, microglial activity, synaptic plasticity, and monoamine metabolism. Its structural stability and dual neuroimmune positioning make Selank a valuable tool in advanced neurobiological research.
Educational & Research Disclaimer
This article is for educational and scientific research purposes only. No therapeutic claims or usage recommendations are provided. Compounds referenced are not approved for human use and are intended solely for controlled laboratory experimentation.
FAQ:
What is Selank?
Selank is a synthetic heptapeptide derived from the endogenous immunomodulatory peptide tuftsin. It has been studied in experimental models for its role in neuromodulatory signaling and immune–neural communication.
How is Selank classified in research literature?
In the scientific literature, Selank is classified as a neuromodulatory peptide and is primarily investigated in preclinical and mechanistic research models rather than clinical therapeutic contexts.
What biological pathways are studied with Selank?
Research involving Selank has examined pathways related to neurotransmitter regulation, stress-response signaling, immune–neural interactions, and BDNF-associated neuromodulatory networks.
Is Selank intended for human or clinical use?
No. Selank is supplied strictly for laboratory and research use. It is not approved for human consumption, medical treatment, or clinical application.
PMID:
- PMID: 10470086 – Experimental studies describing the anxiolytic-like and neuromodulatory properties of Selank in animal models.
- PMID: 16127768 – Research examining Selank’s influence on neurochemical signaling and stress-related pathways in preclinical systems.
- PMID: 12617360 – Analysis of tuftsin-derived peptides, including Selank, and their immunomodulatory and neuroimmune interactions.
Related Searches:
Dihexa — Neurotrophic Peptide Research Article (Educational • Research Use Only)
Semax : ACTH(4–10)-Derived Heptapeptide and Neurotrophic Research Pathways

Selank 10mg
Selank 10mg is a research compound studied for neuroregulatory signaling, stress-response modulation, and anxiolytic pathway mechanisms. For research use only.

Introduction
Tesofensine is a synthetic tropane derivative originally investigated for neurodegenerative disorders and later studied for its impact on metabolic regulation, appetite control, and energy expenditure. It functions primarily as a triple monoamine reuptake inhibitor, influencing dopamine, norepinephrine, and serotonin signaling. Tesofensine has become a molecule of interest in research exploring central energy balance, neuroendocrine signaling, and metabolic homeostasis.
Molecular Mechanism of Action
Tesofensine inhibits presynaptic reuptake transporters for dopamine (DAT), norepinephrine (NET), and serotonin (SERT), resulting in elevated synaptic concentrations of these neurotransmitters. This mechanism enhances catecholaminergic and serotonergic signaling in brain regions responsible for reward, appetite, and energy regulation. Research models indicate that these effects influence hypothalamic control of hunger and thermogenic pathways.
Central Nervous System and Hypothalamic Pathways
Research into Tesofensine focuses heavily on its modulation of hypothalamic signaling networks governing food intake and energy balance. Elevated monoamine activity in the arcuate nucleus and paraventricular nucleus impacts neuropeptides such as neuropeptide Y (NPY), agouti-related peptide (AgRP), and proopiomelanocortin (POMC), resulting in altered appetite signaling and metabolic rate adjustments.
Metabolic and Energy Expenditure Research
Tesofensine is studied for its effects on basal metabolic rate, lipid oxidation, and mitochondrial energy metabolism. Increased sympathetic nervous system activity leads to elevated thermogenesis and enhanced fatty acid utilization. Studies explore its role in modifying energy partitioning and substrate preference in skeletal muscle and adipose tissue.
Neuroendocrine and Peripheral Integration
Beyond its central actions, Tesofensine influences systemic metabolic processes through neuroendocrine cross-talk. Research suggests altered signaling in the hypothalamic–pituitary–adrenal (HPA) and thyroid axes, contributing to modulation of metabolic hormones such as leptin, insulin, and thyroid hormones. These interactions are being examined for their relevance to weight regulation and metabolic adaptation.
Cardiometabolic and Sympathetic Activity
Due to its sympathomimetic effects, Tesofensine research includes analysis of cardiovascular responses such as increased heart rate, blood pressure modulation, and vascular tone. Studies investigate mechanisms for balancing enhanced metabolic rate with cardiovascular safety in controlled models.
Combination and Comparative Research
Recent research explores combination models of Tesofensine with other metabolic regulators, including GLP-1 receptor agonists, AMPK activators, and mitochondrial peptides. Such pairings are designed to examine additive or synergistic effects on energy metabolism, insulin sensitivity, and appetite regulation.
Summary
Tesofensine is a triple monoamine reuptake inhibitor studied for its influence on appetite regulation, energy expenditure, and neuroendocrine signaling. Its integrated actions across central and peripheral systems make it a versatile model compound for investigating the molecular and neural basis of metabolic control in research settings.
Educational & Research Disclaimer
This article is for educational and scientific research purposes only. No therapeutic claims or usage recommendations are provided. Compounds referenced are not approved for human use and are intended solely for controlled laboratory experimentation.
FAQ:
What is Tesofensine?
Tesofensine is a synthetic tropane derivative studied in research for its role as a triple monoamine reuptake inhibitor, affecting dopamine, norepinephrine, and serotonin signaling pathways.
How does Tesofensine work at the molecular level?
Tesofensine inhibits the reuptake transporters for dopamine (DAT), norepinephrine (NET), and serotonin (SERT), leading to prolonged synaptic availability of these neurotransmitters and altered neuroendocrine signaling.
Why is Tesofensine studied in metabolic research?
Although its primary action is central nervous system signaling, Tesofensine is used in research to examine how central monoamine modulation influences appetite regulation, energy expenditure, and metabolic controlthrough neuroendocrine pathways.
What neurotransmitter systems are involved?
Tesofensine primarily affects dopaminergic, noradrenergic, and serotonergic systems, which are key regulators of motivation, reward processing, autonomic output, and hypothalamic energy balance signaling.
Is Tesofensine a peptide?
No. Tesofensine is not a peptide. It is a small-molecule compound derived from the tropane class.
What research models are used to study Tesofensine?
Tesofensine has been evaluated in cellular assays and animal models to study monoamine transport, neuroendocrine regulation, and centrally mediated metabolic signaling.
Is Tesofensine approved for clinical use?
Tesofensine is primarily a research compound and is not approved for general clinical use. Its applications remain investigational.
Selected References (PMIDs)
- PMID: 16362095 – Tesofensine as a triple monoamine reuptake inhibitor
- PMID: 18248663 – Central monoamine signaling and appetite regulation
- PMID: 19351912 – Neuroendocrine control of energy balance via monoamines
- PMID: 20479941 – Dopamine and norepinephrine pathways in metabolic regulation
- PMID: 21407119 – Serotonergic signaling and hypothalamic energy homeostasis
- PMID: 23183208 – CNS-mediated mechanisms linking neurotransmitters and metabolism
RELATED SEARCHES:
Dihexa — Neurotrophic Peptide Research Article (Educational • Research Use Only)
Semax : ACTH(4–10)-Derived Heptapeptide and Neurotrophic Research Pathways
Tesofensine 500mcg (100 ct)
Tesofensine 500mcg is a research compound studied for monoamine transporter modulation, central appetite signaling, and energy balance regulation. For research use only.
