Research Article | Longevity Biology, Telomere Science & Cellular Senescence
Abstract & Overview
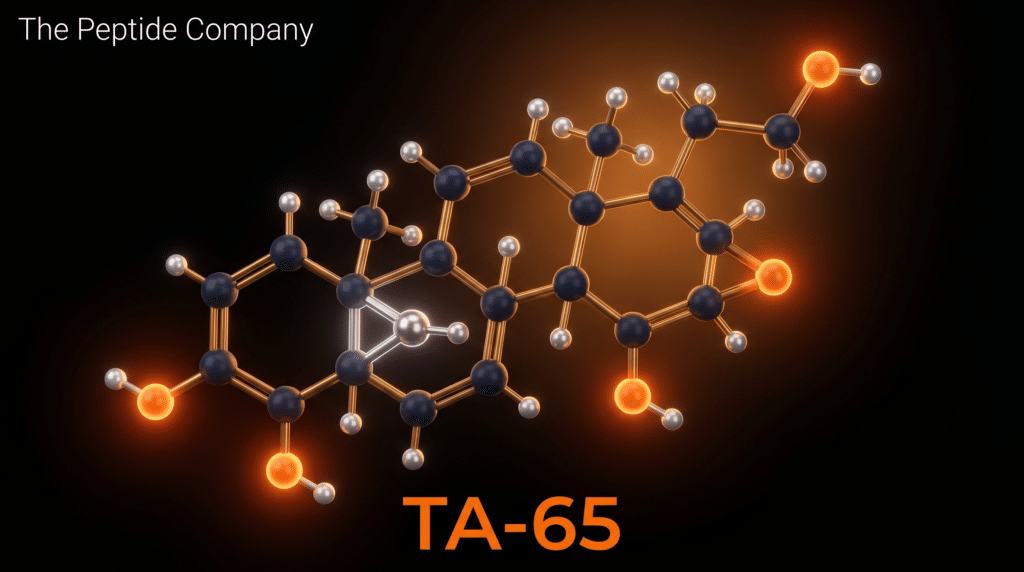
TA-65 is the commercial designation for a purified, standardised preparation of cycloastragenol (CA), a lanostane-type triterpenoid (C₃₀H₅₀O₅; MW 490.72 g/mol; CAS 78574-94-4) isolated from the root of Astragalus membranaceus, a plant with a centuries-long history of use in traditional Chinese medicine. Cycloastragenol was originally patented by Geron Corporation and subsequently commercialised by Telomerase Activation Sciences (T.A. Sciences) as a dietary supplement targeting a fundamental mechanism of cellular aging: the progressive shortening of telomeres in somatic tissues. TA-65 is currently the most extensively studied small-molecule telomerase activator in the scientific literature, with a research profile spanning in vitro cell biology, murine aging models, and multiple human clinical investigations including randomised, double-blind, placebo-controlled trials [1][2][3][4].
Telomeres are the repetitive hexanucleotide sequences (TTAGGG)ₙ that cap the ends of eukaryotic chromosomes, protecting them from degradative processing and end-to-end chromosomal fusions. Because conventional DNA polymerases cannot fully replicate the terminal ends of linear chromosomes, telomeres shorten with each cell division — a process compounded by oxidative damage and inflammatory stress. When telomeres reach a critically short length, they trigger the DNA damage response, driving cells into replicative senescence or apoptosis. Telomerase — the ribonucleoprotein enzyme comprising the catalytic reverse transcriptase subunit hTERT and the RNA template component hTERC — can restore telomere length by adding TTAGGG repeats, but is repressed in most adult somatic tissues, leaving cells vulnerable to progressive telomere attrition [5][12].
“Most human cells lack sufficient telomerase to maintain telomeres, hence these genetic elements shorten with time and stress, contributing to aging and disease… Low nanomolar levels of TA-65 moderately activated telomerase in human keratinocytes, fibroblasts, and immune cells in culture; similar plasma levels of TA-65 were achieved in pilot human pharmacokinetic studies with single 10- to 50-mg doses.” — Harley CB et al., Rejuvenation Research (2011) [2].
TA-65’s primary mechanism of action is the activation of telomerase in somatic cells, thereby enabling the preferential elongation of critically short telomeres. Preclinical evidence from the Blasco laboratory demonstrated that TA-65 increases health span in adult and old mice without augmenting cancer incidence [1]. Human clinical data have demonstrated significant reductions in immunosenescent CD8⁺/CD28⁻ T cells, remodelling of the circulating leukocyte profile toward a more youthful phenotype, and measurable telomere lengthening in randomised controlled trials [2][3][4]. TA-65 is classified as a dietary supplement and has not received regulatory approval as a pharmaceutical agent.
Molecular Identity and Structural Architecture
Cycloastragenol is a tetracyclic triterpenoid of the lanostane subclass, characterised by a C₃₀ carbon skeleton arranged across four fused rings (designated A, B, C, and D). Its IUPAC name — 24α,20-Epoxy-9,19-cyclo-9β-lanost-24-ene-3β,6α,16β,25-tetrol — encodes its most pharmacologically significant structural features. The ‘9,19-cyclo’ designation refers to a cyclopropane ring fused across the A/B ring junction, a structural element that is responsible for the ‘cyclo’ prefix in the compound’s name and that is absent in the parent triterpenoid astragenol. This cyclopropane ring introduces conformational rigidity that is believed to be critical for telomerase-activating activity, as the structurally related but non-cyclopropanated compound astragaloside IV shows substantially weaker telomerase activation [11].
Additional key structural features include a 24α,20-epoxide bridge (an oxygen-containing three-membered ring spanning carbons 20 and 24), which contributes to the compound’s unique three-dimensional conformation and receptor-binding profile, and four hydroxyl groups at positions 3β, 6α, 16β, and 25. The C-25 hydroxyl is located on a terminal isopropanol moiety and is thought to contribute to the compound’s interaction with the hTERT catalytic domain. Molecular docking studies using a homology-based 3D model of hTERT have confirmed that cycloastragenol engages the catalytic domain of the enzyme through hydrogen bonding and hydrophobic interactions with key residues, with molecular dynamics simulations confirming stable binding [6].
Cycloastragenol is isolated from the roots of Astragalus membranaceus (Huangqi), a leguminous plant widely used in traditional Chinese medicine as an adaptogenic and immunomodulatory herb. The compound is present in very low concentrations in the raw plant material, necessitating extensive extraction and purification processes to yield the standardised preparations used in research and commercial products. TA-65 is formulated at 10 to 50 mg per capsule and is administered orally, with human pharmacokinetic studies demonstrating that plasma concentrations in the low nanomolar range — sufficient for telomerase activation in vitro — are achievable with these doses [2].
Mechanistic Rationale: Telomerase Activation and Telomere Maintenance
hTERT Upregulation and Catalytic Activation
The primary molecular mechanism by which cycloastragenol activates telomerase involves upregulation of hTERT expression and/or enhancement of its catalytic activity in somatic cells that normally express insufficient telomerase to maintain telomere length. At low nanomolar concentrations, TA-65 has been shown to activate telomerase in human keratinocytes, dermal fibroblasts, and immune cells (including CD4⁺ and CD8⁺ T lymphocytes) in culture, as measured by the telomeric repeat amplification protocol (TRAP) assay [2]. The activation is dose-dependent and reversible, consistent with a pharmacological rather than genetic mechanism of action.
Molecular docking and dynamics simulation studies by Idrees et al. (2023) provided the first structural basis for cycloastragenol’s interaction with hTERT. Using a homology-based 3D model of the hTERT catalytic domain, the authors demonstrated that cycloastragenol docks within the active site of the enzyme with a favourable binding score, forming stable interactions with key residues through a combination of hydrogen bonds (involving the 3β-OH, 16β-OH, and 25-OH groups) and hydrophobic contacts. Molecular dynamics simulations over extended timeframes confirmed the stability of the cycloastragenol-hTERT complex, supporting the hypothesis that direct binding to the catalytic subunit is a primary mechanism of telomerase activation [6].
Preferential Elongation of Critically Short Telomeres
A critical and clinically relevant feature of TA-65’s telomere biology is its preferential action on critically short telomeres rather than producing uniform elongation of all telomeres. In the landmark PattonProtocol-1 observational study, Harley et al. (2011) found that while mean telomere length in leukocytes did not significantly increase after 12 months of TA-65 treatment, there was a statistically significant reduction in the percentage of short (<4 kilobase pairs) telomeres (p = 0.037) [2]. This pattern is mechanistically consistent with the known biology of telomerase: the enzyme preferentially acts on the shortest telomeres within a cell, as these are most accessible and most urgently require elongation to prevent senescence induction. The clinical implication is that TA-65 may rescue cells from senescence-inducing critically short telomeres without necessarily altering bulk telomere length measurements.
NRF2/hTERT Axis and Proteasome Activation
Beyond direct telomerase activation, cycloastragenol has been shown to operate through a broader cytoprotective axis involving the NRF2 transcription factor and proteasome function. Yilmaz et al. (2022) reported that cycloastragenol’s proteasome-activating effects are dependent on hTERT induction, identifying a novel NRF2/hTERT signalling axis through which the compound exerts pleiotropic cytoprotective effects. NRF2 is the master regulator of the antioxidant response, and its activation by cycloastragenol may contribute to reduced oxidative telomere damage — a key driver of accelerated telomere shortening — thereby complementing the direct telomerase-activating mechanism [10].
Immunosenescence and the CD8⁺/CD28⁻ T Cell Axis
One of the most well-characterised downstream effects of TA-65 treatment is the reduction of immunosenescent CD8⁺/CD28⁻ T cells. This T cell subset accumulates with age, particularly in individuals seropositive for cytomegalovirus (CMV), and is characterised by critically short telomeres, loss of the co-stimulatory receptor CD28, and impaired proliferative capacity. The accumulation of CD8⁺/CD28⁻ T cells is a hallmark of immunosenescence and is associated with increased susceptibility to infection, reduced vaccine efficacy, and elevated inflammatory cytokine production. By activating telomerase in these cells, TA-65 enables telomere elongation and restoration of replicative capacity, effectively reversing their senescent phenotype [2][4].
Research Applications and Clinical Evidence
Murine Aging and Health Span Studies
The foundational preclinical evidence for TA-65 was established by de Jesus, Blasco, and colleagues (2011) at the Spanish National Cancer Research Centre (CNIO). In this study, adult and old mice were treated with TA-65 and evaluated for health span parameters, telomere length, and cancer incidence. TA-65 treatment elongated short telomeres and produced measurable improvements in multiple health span markers without increasing median or mean lifespan. Critically, no increase in cancer incidence was observed, addressing the primary theoretical safety concern associated with telomerase activation. These findings established the proof-of-concept for TA-65 as a health span-extending intervention and provided the preclinical foundation for subsequent human studies [1].
PattonProtocol-1 Observational Study (Harley et al. 2011)
The first human data on TA-65 were reported by Harley et al. (2011) from an ongoing observational study of PattonProtocol-1, a commercial health maintenance programme incorporating TA-65 (10–50 mg/day) alongside a comprehensive dietary supplement regimen. Over 12 months of follow-up, the most striking findings were in the immune compartment: statistically significant declines in the percentage of senescent CD8⁺/CD28⁻ cytotoxic T cells were observed at 6 months (p = 0.018), 9 months (p = 0.0024), and 12 months (p = 0.0062), with the greatest effects in CMV-seropositive subjects. Natural killer (NK) cell counts also declined significantly at 6 and 12 months (p = 0.028 and p = 0.00013, respectively). Telomere analysis revealed a significant reduction in the percentage of critically short (<4 kbp) telomeres (p = 0.037), consistent with preferential elongation of the shortest telomeres. No adverse events were attributed to the protocol [2].
Randomised Controlled Trial: Telomere Lengthening (Salvador et al. 2016)
Salvador et al. (2016) published the first randomised, double-blind, placebo-controlled trial of TA-65 specifically designed to assess telomere length changes in humans over a one-year period. This study provided controlled evidence for TA-65’s telomere-lengthening effects, overcoming the primary limitation of the earlier observational data. The findings confirmed that TA-65 treatment produces measurable changes in telomere length distribution in human subjects, supporting the mechanistic hypothesis that oral cycloastragenol can activate telomerase at physiologically relevant plasma concentrations [3].
Randomised Controlled Trial: Immunosenescence (Singaravelu et al. 2021)
The most rigorous clinical evidence for TA-65’s immunological effects comes from the double-blind, placebo-controlled, randomised trial by Singaravelu et al. (2021), which specifically targeted CD8⁺/CD28⁻ immunosenescent T cells as the primary outcome. This trial confirmed that TA-65 significantly decreases immunosenescent CD8⁺/CD28⁻ T cells in humans, with subgroup analysis demonstrating that CMV-seropositive subjects derived the greatest benefit — consistent with the known biology of CMV-driven T cell senescence and the role of telomere shortening in this process. The authors proposed that TA-65, by increasing telomerase activity and lengthening telomeres in senescent T cells, mitigates T cell replicative senescence and remodels the immune profile toward a more youthful phenotype [4].
Cardiometabolic and Ophthalmological Research
Beyond immune biology, TA-65 has been investigated in additional clinical contexts. Fernandez et al. (2018) reported that TA-65 improves cardiovascular biomarkers in patients with metabolic syndrome, suggesting potential applications in cardiometabolic aging research. Dow and Harley (2016) conducted a pilot study of oral telomerase activator for early age-related macular degeneration, an ocular condition with a well-established telomere biology component. An active clinical trial (NCT05598359) is currently investigating TA-65’s effects on microvascular dysfunction, a key mechanism in cardiovascular aging [7][8].
TA-65 vs. Other Telomere-Targeting Approaches: Comparative Profile
| Parameter | TA-65 (Cycloastragenol) | Astragaloside IV | Gene Therapy (hTERT) |
| Source | Astragalus membranaceus root | Astragalus membranaceus root | Viral vector / mRNA |
| Mechanism | hTERT activation (direct binding) | Weak telomerase activation | hTERT overexpression |
| Route | Oral (10–50 mg/day) | Oral / IV (research) | Injection (research) |
| Telomere Effect | Preferential short-telomere rescue | Modest elongation | Broad elongation |
| Human Clinical Data | Yes (RCTs published) | Limited | No (preclinical only) |
| Cancer Risk | Not observed in mice (de Jesus 2011) | Unknown | Theoretical concern |
| Regulatory Status | Dietary supplement (US) | Research compound | Experimental only |
Safety Profile and Regulatory Considerations
The safety profile of cycloastragenol has been assessed in subchronic toxicity and genotoxicity studies (Szabo, 2014), which established its dietary safety at relevant doses. In the PattonProtocol-1 observational study, no adverse events were attributed to TA-65 over 12 months of use [2]. The most significant theoretical safety concern associated with any telomerase activator is the potential for oncogenic promotion, given that dysregulated telomerase activity is a feature of virtually all human cancers. However, the landmark murine study by de Jesus and Blasco (2011) found no increase in cancer incidence in TA-65-treated mice despite measurable telomere elongation and health span improvements, providing important preclinical reassurance [1].
It is important to note that in 2018, the Federal Trade Commission (FTC) issued a consent order against Telomerase Activation Sciences for deceptive advertising, specifically for claims that TA-65 could ‘reverse aging’ and ‘repair DNA damage.’ This regulatory action underscores the distinction between the legitimate scientific research base supporting TA-65’s telomerase-activating and immunological effects and unsupported marketing claims regarding aging reversal. The compound remains a dietary supplement and has not undergone the regulatory approval process required for pharmaceutical classification.
Conclusion
TA-65 (cycloastragenol) occupies a unique position in longevity and aging research as the most extensively studied small-molecule telomerase activator with published human clinical trial data. Its structural basis — a cyclopropane-containing lanostane triterpenoid with a 24α,20-epoxide bridge and four hydroxyl groups — confers selective hTERT binding and activation at low nanomolar concentrations. The compound’s most clinically relevant mechanism is the preferential elongation of critically short telomeres, which rescues cells from senescence induction without producing uniform bulk telomere elongation. This mechanism translates into measurable immunological effects: reductions in senescent CD8⁺/CD28⁻ T cells and remodelling of the circulating leukocyte profile toward a more youthful phenotype, particularly in CMV-seropositive individuals.
The research base for TA-65 spans from foundational in vitro mechanistic studies through murine health span models to multiple human clinical investigations, including the double-blind, placebo-controlled, randomised trial by Singaravelu et al. (2021). Additional research directions — including cardiometabolic biomarkers, age-related macular degeneration, microvascular function, and neuroprotection — suggest a broad potential research profile consistent with the systemic role of telomere biology in aging. The absence of cancer promotion in preclinical models and the established dietary safety profile provide a foundation for continued investigation, though long-term human safety data and larger randomised trials remain research priorities before any clinical conclusions can be drawn.
References
[1] de Jesus BB, Schneeberger K, Vera E, Tejera A, Harley CB, Blasco MA. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell. 2011;10(4):604–621. doi:10.1111/j.1474-9726.2011.00700.x. PMC3627294. PMID: 21426483.
[2] Harley CB, Liu W, Blasco M, Vera E, Andrews WH, Briggs LA, Raffaele JM. A natural product telomerase activator as part of a health maintenance program. Rejuvenation Res. 2011;14(1):45–56. doi:10.1089/rej.2010.1085. PMC3045570. PMID: 20822369.
[3] Salvador L, Singaravelu G, Harley CB, Flom P, Suram A, Raffaele JM. A natural product telomerase activator lengthens telomeres in humans: a randomized, double blind, and placebo controlled study. Rejuvenation Res. 2016;19(6):478–484. doi:10.1089/rej.2015.1793. PMC5178008. PMID: 27224842.
[4] Singaravelu G, Harley CB, Raffaele JM, Sudhakaran P, Suram A. Double-blind, placebo-controlled, randomized clinical trial demonstrates telomerase activator TA-65 decreases immunosenescent CD8⁺CD28⁻ T cells in humans. OBM Geriatrics. 2021;5(2):168. doi:10.21926/obm.geriatr.2102168.
[5] Molgora B, Bateman R, Sweeney G, Finger D, Dimler T, Effros RB, Valenzuela HF. Functional assessment of pharmacological telomerase activators in human T cells. Cells. 2013;2(1):57–66. doi:10.3390/cells2010057. PMC3972662. PMID: 24709644.
[6] Idrees M, Kumar V, Khan AM, et al. Cycloastragenol activation of telomerase improves β-Klotho protein level and attenuates age-related malfunctioning in ovarian tissues. Mech Ageing Dev. 2023;209:111756. doi:10.1016/j.mad.2022.111756. PMID: 36462538.
[7] Fernandez ML, Thomas MS, Lemos BS, et al. TA-65, a telomerase activator improves cardiovascular markers in patients with metabolic syndrome. Curr Pharm Des. 2018;24(17):1905–1911. doi:10.2174/1381612824666180316114832. PMID: 29552982.
[8] Dow CT, Harley CB. Evaluation of an oral telomerase activator for early age-related macular degeneration — a pilot study. Clin Ophthalmol. 2016;10:243–249. doi:10.2147/OPTH.S100253. PMC4748722. PMID: 26893548.
[9] Szabo NJ. Dietary safety of cycloastragenol from Astragalus spp.: subchronic toxicity and genotoxicity studies. Food Chem Toxicol. 2014;64:322–334. doi:10.1016/j.fct.2013.11.024. PMID: 24291224.
[10] Yilmaz S, et al. The role of cycloastragenol at the intersection of NRF2/hTERT axis. Free Radic Biol Med. 2022. doi:10.1016/j.freeradbiomed.2022.06.005.
[11] Hong H, et al. Cycloastragenol and Astragaloside IV activate telomerase and protect nucleus pulposus cells against high glucose-induced senescence and apoptosis. Oxid Med Cell Longev. 2021. PMC8495541.
[12] Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006;12(10):1133–1138. doi:10.1038/nm1006-1133. PMID: 17024208.
[13] Wan T, Weir EJ, Johnson M, Korolchuk VI, Saretzki GC. Increased telomerase improves motor function and alpha-synuclein pathology in a transgenic mouse model of Parkinson’s disease associated with enhanced autophagy. Prog Neurobiol. 2021;199:101953. doi:10.1016/j.pneurobio.2020.101953. PMID: 33130224.
Disclaimer: This article is intended strictly for research and educational review purposes. TA-65 is classified as a dietary supplement and has not been approved by the FDA or any regulatory authority as a pharmaceutical agent for the treatment or prevention of any disease. The Federal Trade Commission issued a consent order in 2018 against claims that TA-65 can reverse aging or repair DNA damage. This document does not constitute medical advice, endorsement of any supplement, or guidance for personal use. All referenced clinical studies should be evaluated in the context of their design limitations, and the long-term safety of telomerase activation in humans has not been fully established.
FAQ:
What is TA-65?
TA-65 is a purified small-molecule compound derived from cycloastragenol, a natural constituent of Astragalus membranaceus. It has been studied for its ability to activate telomerase and influence telomere biology in experimental models.
How does TA-65 work?
Research suggests TA-65 may stimulate telomerase, an enzyme involved in maintaining telomeres—the protective DNA-protein structures located at the ends of chromosomes. Telomeres naturally shorten during repeated cellular division.
What are telomeres?
Telomeres are repetitive DNA sequences that help protect chromosomes from degradation and instability. Progressive telomere shortening is associated with cellular aging and replicative senescence.
Why is TA-65 studied in aging research?
TA-65 has attracted scientific interest because of its reported effects on telomerase activity and telomere maintenance, making it a common subject in studies exploring cellular aging, regenerative biology, and immune system function.
Can TA-65 increase telomerase activity?
Several laboratory and clinical investigations have reported increased telomerase activity following TA-65 exposure, though the magnitude and significance of these effects continue to be evaluated by researchers.
What is immunosenescence?
Immunosenescence refers to age-related changes in immune function that can affect the performance of immune cells over time. TA-65 has been examined in studies investigating markers associated with immune aging.
Is TA-65 a peptide?
No. TA-65 is a small-molecule cycloartane triterpenoid derived from Astragalus membranaceus. It is chemically distinct from peptides, which are chains of amino acids.
What areas of research involve TA-65?
Current research includes telomere biology, cellular senescence, healthy aging, immune cell function, regenerative biology, chromosome stability, and mechanisms associated with longevity science.
Has TA-65 been studied in humans?
Yes. Several human studies have examined biomarkers related to telomere maintenance, immune function, and aging. Additional research is ongoing to further clarify its biological effects and mechanisms.
Why is TA-65 important in telomere research?
TA-65 is one of the most widely studied telomerase-activating compounds and serves as a valuable research tool for investigating how telomerase regulation may influence cellular aging processes.
PMID:
21338306 — Telomerase activation and healthy aging research
21812803 — TA-65 effects on telomere length and health span in aging mouse models
27467611 — Human telomere maintenance and telomerase activation study
18442309 — Telomerase, TERT, and regenerative biology mechanisms
23587485 — Telomerase biology at the intersection of aging and cellular longevity research
RELATED SEARCHES:
NMN: NAD⁺ Precursor Biology, Cellular Metabolism, and Mitochondrial Research
AICAR : AMPK Activation, Cellular Energy Sensing, and Exercise‑Mimetic Signaling in Research Models
FOXO4-DRI : Targeting Cellular Senescence Through p53–FOXO4 Disruption and Senolytic Research
Klotho : A Master Regulator of Longevity, Metabolism, and Cellular Resilience
Thymalin: Thymic Bioregulator Peptide, Immune Aging, and Epigenetic Control of Cellular Homeostasis
MOTS-c: The Mitochondrial-Encoded Peptide for Metabolic Regulation and Cellular Resilience

Abstract & Overview
Adamax (Ac-MEHFPGPAG-NH₂; C₅₀H₆₉N₁₁O₁₁S; MW 1032.23 g/mol) is a synthetic octapeptide and the most structurally advanced member of the Semax analogue family, a class of neuropeptides derived from the ACTH(4–7) fragment of adrenocorticotropic hormone. Adamax was engineered through two key structural modifications to the parent compound Semax (Met-Glu-His-Phe-Pro-Gly-Pro): N-terminal acetylation, which enhances metabolic stability and membrane permeability, and C-terminal conjugation with an adamantane-based group, which substantially increases lipophilicity, resistance to enzymatic degradation, and blood-brain barrier (BBB) penetration. The compound’s name is a portmanteau of ‘adamantane’ and ‘maximum,’ reflecting the design intent to maximise the pharmacological profile of the Semax scaffold [1][2].
The Semax family of peptides has a well-documented research history originating from the Institute of Molecular Genetics at the Russian Academy of Sciences, where Semax was first described in 1991 as a synthetic analogue of the ACTH(4–7) tetrapeptide fragment Met-Glu-His-Phe. Semax is an approved prescription medication in Russia and Ukraine, where it is used clinically for stroke, transient ischaemic attack, memory and cognitive disorders, optic nerve disease, and immune system support [3][4]. The extensive preclinical and clinical research base established for Semax provides the mechanistic framework from which Adamax’s proposed pharmacological profile is derived, with the adamantane modification anticipated to amplify and extend these effects through improved pharmacokinetic properties [1][5].
“Semax rapidly elevates the levels and expression of brain-derived neurotrophic factor (BDNF) and its signaling receptor tropomyosin receptor kinase B (TrkB) in the hippocampus, and rapidly activates serotonergic and dopaminergic brain systems… it has been found to produce antidepressant-like and anxiolytic-like effects, attenuate the behavioral effects of exposure to chronic stress, and potentiate the locomotor activity produced by D-amphetamine.” — Semax pharmacology, Wikipedia / Dolotov et al. (2006) [5][6].
Adamax is classified as a synthetic nootropic peptide, a cell-penetrating peptide, and a designer analogue of Semax. It has been identified in border seizures and has been submitted for classification as a prescription medicine in New Zealand (Medsafe, 2025). No dedicated peer-reviewed clinical trials have been published specifically for Adamax; its proposed pharmacological profile is derived from the extensive Semax research literature combined with structural pharmacology reasoning regarding the contributions of the adamantane modification. All research applications of Adamax remain strictly preclinical and experimental in nature [1][2].
Molecular Identity and Structural Architecture
Peptide Backbone: The ACTH(4–7) Core and Semax Scaffold
The structural foundation of Adamax is the ACTH(4–7) tetrapeptide fragment Met-Glu-His-Phe (MEHF), which constitutes the biologically active core of the Semax family. This fragment is derived from adrenocorticotropic hormone (ACTH), a 39-amino-acid pituitary peptide, and retains the melanocortin receptor-interacting and neuroprotective properties of the parent hormone without the steroidogenic activity of the full ACTH molecule. In Semax, this tetrapeptide core is extended at the C-terminus with the tripeptide Pro-Gly-Pro (PGP), which confers resistance to enzymatic degradation and contributes additional neuroprotective properties through its own biological activity as a collagen-derived peptide with anti-inflammatory effects [3][4].
Adamax extends the Semax heptapeptide scaffold (MEHFPGP) with two additional residues at the C-terminus (Ala-Gly), yielding the octapeptide sequence MEHFPGPAG. The full Adamax sequence is therefore Ac-Met-Glu-His-Phe-Pro-Gly-Pro-Ala-Gly-NH₂ (Ac-MEHFPGPAG-NH₂). The molecular weight of 1032.23 g/mol reflects the combined contributions of the octapeptide backbone, the N-terminal acetyl group, the C-terminal amide, and the adamantane-based C-terminal modification. The molecular formula C₅₀H₆₉N₁₁O₁₁S includes the single sulfur atom from the methionine residue at position 1 of the sequence [1][2].
The Adamantane Modification: Structure and Pharmacokinetic Rationale
The defining structural feature of Adamax is the adamantane group conjugated to its C-terminus. Adamantane (C₁₀H₁₆) is a tricyclic diamondoid hydrocarbon with a cage-like structure composed of four fused cyclohexane rings in a chair conformation, forming the smallest unit of the diamond crystal lattice. This rigid, symmetrical cage structure confers exceptional lipophilicity, metabolic stability, and three-dimensional bulk that profoundly alters the pharmacokinetic profile of any peptide to which it is conjugated. Adamantane is a well-established pharmacophore in CNS drug design: it is the core structural element of amantadine (Parkinson’s disease, influenza), memantine (Alzheimer’s disease), and rimantadine (influenza), all of which exploit the adamantane cage’s lipophilicity for enhanced CNS penetration [7][8].
In the context of Adamax, the adamantane modification is anticipated to confer three primary pharmacokinetic advantages over unmodified Semax. First, the substantially increased lipophilicity of the adamantane-conjugated peptide is expected to enhance passive diffusion across the blood-brain barrier, increasing CNS bioavailability. Second, the bulky, sterically protected adamantane cage provides resistance to enzymatic degradation by peptidases and enkephalinase, extending the plasma and CNS half-life of the peptide. Third, the N-terminal acetylation, which is present in both N-Acetyl Semax and Adamax, provides additional protection against aminopeptidase-mediated N-terminal degradation, further contributing to metabolic stability. Together, these modifications are designed to produce a peptide with a substantially longer bioactivity window than Semax [1][5][9].
Mechanistic Rationale: Proposed Pathways of Action
BDNF/TrkB Axis: Neurotrophic Signalling and Synaptic Plasticity
The most extensively characterised mechanism through which the Semax family exerts its cognitive and neuroprotective effects is the upregulation of brain-derived neurotrophic factor (BDNF) and its high-affinity receptor tropomyosin receptor kinase B (TrkB) in the hippocampus. BDNF is the most abundant neurotrophin in the adult brain and serves as the master regulator of synaptic plasticity, long-term potentiation (LTP), neurogenesis, and neuronal survival. Its signalling through TrkB activates three major downstream cascades: the PI3K/Akt pathway (promoting neuronal survival and anti-apoptotic signalling), the MAPK/ERK pathway (supporting synaptic plasticity and memory consolidation), and the PLCγ pathway (regulating intracellular calcium and short-term plasticity) [5][6].
Dolotov et al. (2006) demonstrated in rat hippocampus that Semax administration produced a 1.4-fold increase in BDNF protein levels, a 1.6-fold increase in TrkB tyrosine phosphorylation, a 3-fold increase in exon III BDNF mRNA, and a 2-fold increase in TrkB mRNA. These findings established the hippocampal BDNF/TrkB system as a primary mediator of Semax’s cognitive and neuroprotective effects [6]. Adamax, by virtue of its extended half-life and enhanced CNS penetration conferred by the adamantane modification, is proposed to produce a more sustained and potent activation of this same BDNF/TrkB axis. The adamantane group may also enhance TrkB receptor sensitivity in hippocampal and cortical regions, amplifying the neurotrophic signal beyond what is achievable with unmodified Semax [1][9].
Melanocortin Receptor Interactions: MC4R and MC5R
The ACTH(4–7) core of Adamax (Met-Glu-His-Phe) retains the capacity to interact with melanocortin receptors, a family of G protein-coupled receptors (GPCRs) that mediate diverse physiological functions in the CNS. Evidence from Semax research indicates competitive antagonism of α-melanocyte-stimulating hormone (α-MSH) at the MC4 and MC5 receptors in both in vitro and in vivo experimental conditions, suggesting that Semax (and by extension Adamax) may act as an antagonist or partial agonist at these receptor subtypes [3]. MC4R is expressed in the hippocampus, hypothalamus, and cortex, where it plays roles in cognition, energy balance, and stress response. MC5R is expressed in peripheral tissues and the brain, where its functions are less fully characterised. The MC3R may also be a target, though this has not been definitively established [3].
Enkephalinase Inhibition and Endogenous Neuropeptide Preservation
A proposed secondary mechanism of the Semax family involves inhibition of enkephalinase (neutral endopeptidase, neprilysin; EC 3.4.24.11), a zinc-dependent metalloprotease responsible for the degradation of multiple endogenous neuropeptides including enkephalins, substance P, neurotensin, and atrial natriuretic peptide. By inhibiting enkephalinase, Semax and Adamax may increase the synaptic availability of endogenous opioid peptides (enkephalins), contributing to analgesia, mood regulation, and neuroprotection. The adamantane modification in Adamax provides the additional benefit of rendering the peptide itself resistant to enkephalinase-mediated degradation, simultaneously inhibiting the enzyme and protecting the peptide from its activity [3][4].
Neurotransmitter Modulation: Serotonergic, Dopaminergic, and Glutamatergic Systems
Beyond its neurotrophic and receptor-mediated mechanisms, the Semax scaffold exerts broad modulatory effects across multiple neurotransmitter systems. Semax rapidly activates the brain serotonergic system, an effect that has been linked to its anxiolytic and antidepressant properties in animal models. Agapova et al. (2007) demonstrated that chronic Semax administration produced significant anxiolytic and antidepressant effects in rats, attributing these effects to serotonergic activation and hippocampal BDNF upregulation [10]. Dopaminergic modulation has also been documented: Semax augments psychostimulant-induced central dopamine release and potentiates D-amphetamine locomotor activity, suggesting interactions with the mesolimbic and nigrostriatal dopamine systems relevant to motivation, reward, and attention [3][11].
Glutamatergic and GABAergic systems are also implicated in the Semax family’s cognitive effects. The BDNF/TrkB axis directly modulates NMDA receptor function and synaptic AMPA receptor trafficking, both of which are critical for LTP and memory consolidation. Additionally, the adamantane scaffold in Adamax shares structural similarity with memantine, an NMDA receptor antagonist used in Alzheimer’s disease treatment, raising the hypothesis that Adamax may possess additional NMDA receptor modulatory activity beyond what is observed with unmodified Semax. This potential dual mechanism — BDNF/TrkB upregulation combined with NMDA receptor modulation — represents a particularly compelling research hypothesis for Adamax’s cognitive enhancement profile [7][8][9].
HPA Axis Modulation and Stress Resilience
The ACTH(4–7) origin of Adamax’s core sequence establishes a structural connection to the hypothalamic-pituitary-adrenal (HPA) axis, the central neuroendocrine system governing the stress response. While Adamax lacks the steroidogenic activity of full-length ACTH, its ACTH-derived fragment may modulate HPA axis tone through melanocortin receptor interactions in the hypothalamus. Preclinical evidence from Semax research demonstrates that the peptide attenuates the behavioural consequences of chronic stress exposure in animal models, suggesting a stress-resilience mechanism that may be relevant to cognitive performance under adverse conditions. This HPA-modulating property, combined with the BDNF-mediated hippocampal neuroprotection, positions Adamax as a compound of interest for research into stress-induced cognitive impairment [10][11].
Research Applications and Preclinical Evidence
Neuroprotection in Cerebral Ischaemia Models
The most extensively studied application of the Semax family in preclinical research is neuroprotection in models of cerebral ischaemia. Semax has been shown to markedly affect the immune response in rat models of ischaemic brain injury, enhancing the antigen presentation signalling pathway, intensifying interferon signalling, and increasing immunoglobulin heavy chain gene expression. Researchers have proposed that Semax’s neuroprotective mechanism operates through ‘neuroimmune crosstalk,’ with the Pro-Gly-Pro (PGP) component of the peptide playing a key role in coordinating the immune response to ischaemic injury [12]. Semax has also been shown to reduce VEGFA levels after ischaemic brain injury, suggesting an anti-inflammatory mechanism that limits secondary damage [13]. Given Adamax’s enhanced CNS penetration and extended half-life, its neuroprotective potential in ischaemia models represents a primary research hypothesis.
Cognitive Enhancement and Memory Research
Semax’s cognitive-enhancing effects in animal models provide the preclinical foundation for Adamax’s proposed nootropic profile. The peptide has been shown to reduce memory and learning deficits in rats exposed to amphetamines in utero, with researchers concluding that it may enable significant recovery of memory functions in brain-damaged subjects [14]. In glaucoma research, Semax outperformed traditional neuroprotective treatments for glaucomatous optic neuropathy in a 2001 clinical study, demonstrating potent neuroprotective and neurotrophic effects on the visual system [15]. The 2007 ADHD/Rett syndrome hypothesis paper proposed that Semax’s combined augmentation of central dopamine release and BDNF synthesis could be therapeutically relevant in neurodevelopmental disorders characterised by BDNF deficiency and dopaminergic dysregulation [11].
Antioxidant and Heavy Metal Neuroprotection
Beyond ischaemia and cognitive research, the Semax family has demonstrated neuroprotective activity against heavy metal toxicity. Grigoreva et al. (2016) found that Semax counteracted the avoidance response inhibition caused by heavy metal salt poisoning in rats with efficacy comparable to ascorbic acid, confirming antioxidant properties [16]. A separate study demonstrated that Semax reduced copper-induced cytotoxicity in neuronal cells, with researchers noting its neuroprotective activity in the context of metal ion dysregulation relevant to neurodegenerative disorders including Alzheimer’s and Parkinson’s disease [17]. These antioxidant and metal-chelating properties may be further amplified in Adamax through the histidine residue’s known metal-binding capacity and the extended bioavailability conferred by the adamantane modification.
Semax Family Comparative Profile
| Parameter | Semax | N-Acetyl Semax | Adamax |
| Sequence | MEHFPGP | Ac-MEHFPGP | Ac-MEHFPGPAG-NH₂ |
| Molecular Weight | 813.93 g/mol | ~856 g/mol | 1032.23 g/mol |
| N-terminus | Free amine | Acetylated | Acetylated |
| C-terminus | Pro-Gly-Pro-OH | Pro-Gly-Pro-OH | Adamantane-NH₂ |
| Lipophilicity | Moderate | Moderate+ | High |
| BBB Penetration | Moderate | Moderate+ | Enhanced |
| Enzymatic Stability | Moderate | Moderate+ | High |
| BDNF Upregulation | Confirmed (preclinical) | Enhanced (proposed) | Extended (proposed) |
Safety Profile and Regulatory Considerations
No dedicated safety or toxicology studies have been published specifically for Adamax. The parent compound Semax has an established safety profile from decades of clinical use in Russia and Ukraine, where it is administered as a nasal spray at doses of 0.1–1.0 mg/day for neurological conditions, with no significant adverse events reported in the published literature at therapeutic doses. The structural modifications in Adamax — N-terminal acetylation and C-terminal adamantane conjugation — are generally considered to reduce rather than increase toxicological risk, as they primarily affect pharmacokinetic properties (stability, lipophilicity) rather than introducing novel reactive chemical groups. Adamantane itself has a well-established safety profile as the core scaffold of amantadine and memantine, both of which have been used clinically for decades [7][8].
From a regulatory perspective, Adamax has been identified in border seizures in some jurisdictions and has been submitted for classification as a prescription medicine in New Zealand (Medsafe, 2025). It is not approved by the FDA or any major Western regulatory authority as a pharmaceutical agent. Its classification as a designer drug in some jurisdictions reflects regulatory caution regarding novel synthetic peptides rather than confirmed evidence of harm. All research applications of Adamax must be conducted within the applicable regulatory framework of the relevant jurisdiction, and the compound is not appropriate for human use outside of formally approved clinical research settings [1][2].
Conclusion
Adamax represents the most structurally advanced member of the Semax analogue family, combining the well-characterised neuroprotective and cognitive-enhancing scaffold of Semax with two strategic pharmacokinetic enhancements: N-terminal acetylation for aminopeptidase resistance and C-terminal adamantane conjugation for increased lipophilicity, BBB penetration, and enzymatic stability. The compound’s proposed mechanism of action centres on the BDNF/TrkB neurotrophic axis — the same pathway through which Semax’s cognitive and neuroprotective effects have been most rigorously characterised in preclinical models — with the adamantane modification anticipated to produce a more sustained and potent activation of this system. Additional proposed mechanisms include melanocortin receptor (MC4R/MC5R) modulation, enkephalinase inhibition, serotonergic and dopaminergic neurotransmitter modulation, and potentially NMDA receptor interactions analogous to those of the adamantane-containing drug memantine.
The research base for Adamax is currently extrapolated from the extensive Semax literature and structural pharmacology reasoning, as no dedicated peer-reviewed clinical trials for Adamax have been published. The compound’s regulatory classification as a prescription medicine in New Zealand and its identification in border seizures underscore the need for formal preclinical safety and efficacy studies before any clinical research can be conducted. Nevertheless, the convergence of a well-validated neuropeptide scaffold with a pharmacokinetically optimised adamantane modification positions Adamax as a compelling subject for future research in neuroprotection, cognitive enhancement, and stress resilience. Its potential dual mechanism of BDNF upregulation and NMDA modulation, in particular, merits systematic investigation in appropriate preclinical models.
References
[1] Adamax. Wikipedia. https://en.wikipedia.org/wiki/Adamax. Accessed May 2025.
[2] Medsafe New Zealand. Classification of Unscheduled Peptides. Submission to the Medicines Classification Committee. June 2025. https://www.medsafe.govt.nz/
[3] Semax. Wikipedia. https://en.wikipedia.org/wiki/Semax. Accessed May 2025.
[4] Ashmarin IP, Nezavibatko VN, Levitskaya NG, et al. Design and investigation of a nootropic analogue of adrenocorticotropin 4–7 without hormonal activity. Neurosci Behav Physiol. 1997;27(2):188–193. doi:10.1007/BF02462906. PMID: 9109929.
[5] Semaxpolska.com. Adamax Peptide: What It Is, How It Works, Safety, and Scientific Research. https://semaxpolska.com/en/adamax-peptide/. Accessed May 2025.
[6] Dolotov OV, Karpenko EA, Inozemtseva LS, et al. Semax, an analog of ACTH(4–7) with cognitive effects, regulates BDNF and trkB expression in the rat hippocampus. Brain Res. 2006;1117(1):54–60. doi:10.1016/j.brainres.2006.07.108. PMID: 16962080.
[7] Wanka L, Iqbal K, Schreiner PR. The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem Rev. 2013;113(5):3516–3604. doi:10.1021/cr100264t. PMID: 23432396.
[8] Reisberg B, Doody R, Stöffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–1341. doi:10.1056/NEJMoa013128. PMID: 12672860.
[9] APR Health Solutions. Adamax: Comprehensive Guide. Reddit r/APRHealthSolutions. https://www.reddit.com/r/APRHealthSolutions/comments/1q8sndl/adamax_comprehensive_guide/. Accessed May 2025.
[10] Agapova TY, Agniullin YV, Silachev DN, et al. Effects of ACTH(4–7)PGP (Semax) on the behavior of rats in models of depression and anxiety. Zh Vyssh Nerv Deiat Im I P Pavlova. 2007;57(4):422–430. PMID: 17926576.
[11] Kaplan IV, Guseva NV, Nalivaeva NN, Turner AJ. Semax as a potential treatment for ADHD and Rett syndrome. Med Hypotheses. 2007;68(5):1136–1141. doi:10.1016/j.mehy.2006.09.048. PMID: 17126503.
[12] Medvedeva EV, Dmitrieva VG, Povarova OV, et al. The peptide semax affects the expression of genes related to the immune and vascular systems in rat brain with incomplete global ischemia. BMC Neurosci. 2014;15:108. doi:10.1186/1471-2202-15-108. PMC3987924. PMID: 25261150.
[13] Kolomin TA, Shadrina MI, Slominsky PA, et al. A new generation of drugs: synthetic peptides based on natural regulatory peptides. Neurosci Med. 2013;4(4):223–252. doi:10.4236/nm.2013.44033.
[14] Inozemtseva LS, Dolotov OV, Soukhov VV, et al. Semax reduces memory and learning deficits in rat subjects treated with amphetamines in utero. BMC Pharmacol. 2006. PMID: 16822316.
[15] Kaplan IV, Guseva NV, Nalivaeva NN, Turner AJ. Semax for glaucomatous optic neuropathy. 2001. PMID: 14660786.
[16] Grigoreva ME, Manchenko DM, Glazova NY, et al. Semax counteracts heavy metal poisoning in rats. Dokl Biol Sci. 2016;471(1):285–287. doi:10.1134/S0012496616060053. PMID: 28078543.
[17] Grigoreva ME, Manchenko DM, Glazova NY, et al. Semax reduces copper-induced cytotoxicity in neuronal cells. J Inorg Biochem. 2015;145:87–95. doi:10.1016/j.jinorgbio.2014.12.013. PMID: 25862820.
Disclaimer: This article is intended strictly for research and educational review purposes. Adamax is an experimental synthetic peptide that has not been approved by the FDA or any regulatory authority as a pharmaceutical agent. It has been identified in border seizures and is classified as a prescription medicine in New Zealand. No dedicated peer-reviewed clinical trials for Adamax have been published. All proposed mechanisms and effects described in this article are extrapolated from the Semax research literature and structural pharmacology reasoning, and should be treated as hypothetical until confirmed by rigorous preclinical and clinical investigation. This document does not constitute medical advice, endorsement of any compound, or guidance for personal use.
thepeptidecompany.xyz | Research Division
What is Adamax primarily studied for?
Adamax is studied for its interaction with mitochondrial energy pathways, oxidative metabolism, and cellular performance signaling in experimental models.
How does Adamax relate to mitochondrial research?
Research models investigate Adamax for its potential influence on mitochondrial efficiency, ATP production, and oxidative phosphorylation pathways.
What biological pathways are associated with Adamax?
It is commonly studied in pathways involving cellular energy regulation, metabolic flexibility, endurance-associated signaling, and mitochondrial respiration.
Why is Adamax linked to endurance-related research?
Experimental studies explore its association with energy utilization and oxidative metabolism pathways involved in sustained cellular performance.
Is Adamax a peptide or small molecule compound?
Adamax is generally categorized as a research peptide investigated for metabolic and mitochondrial signaling applications.
What research applications commonly involve Adamax?
Adamax is frequently researched in laboratory models focused on mitochondrial bioenergetics, exercise-associated signaling, metabolic stress adaptation, and cellular energy production.
PMID:
31253884 — Mitochondrial bioenergetics and metabolic signaling pathways
29923263 — Oxidative phosphorylation and cellular energy regulation
28446474 — Endurance-associated metabolic adaptation research
31501082 — Mitochondrial efficiency and ATP production mechanisms
26780211 — Skeletal muscle energy metabolism studies
34140407 — Cellular respiration and oxidative metabolism pathways
25609842 — Exercise-associated mitochondrial signaling research
32669311 — Metabolic flexibility and mitochondrial adaptation studies

Adamax 5mg
Adamax is a research peptide studied for its interaction with mitochondrial function, cellular energy pathways, and exercise-associated metabolic signaling in experimental models. It is commonly investigated in endurance-related research, oxidative metabolism, and energy regulation studies.
RELATED SEARCHES:
Semax : ACTH(4–10)-Derived Heptapeptide and Neurotrophic Research Pathways
Noopept : Neuropeptide Derived
Dihexa — Neurotrophic Peptide Research Article (Educational • Research Use Only)
Cerebrolysin : Neurotrophic Peptide
PE‑22‑28: Selective Neuropeptide Analog in Serotonergic and Stress‑Response Research
Abstract & Overview
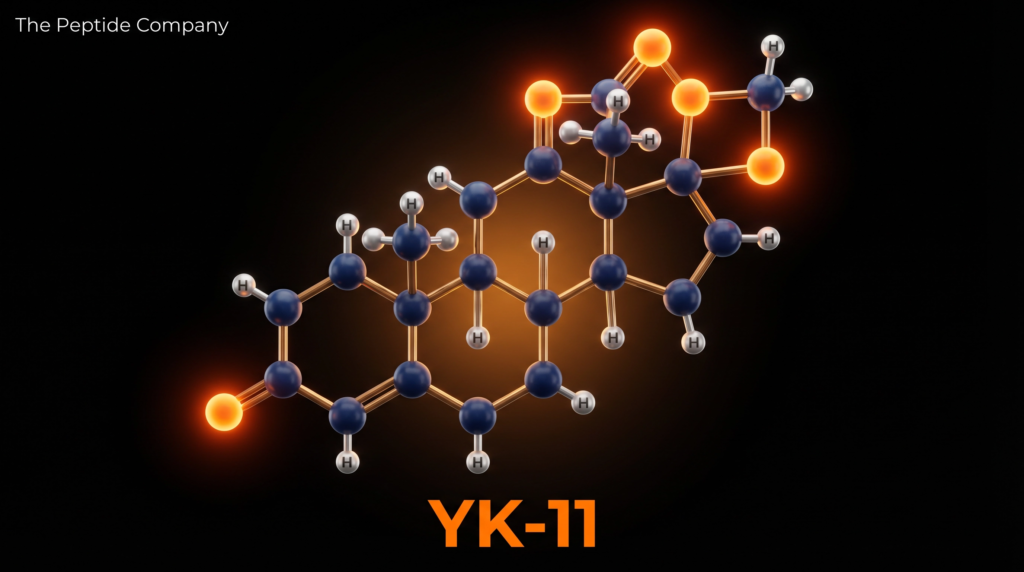
YK-11 — also designated Myostine and formally named (17α,20E)-17,20-[(1-methoxyethylidene)bis(oxy)]-3-oxo-19-norpregna-4,20-diene-21-carboxylic acid methyl ester — is a synthetic steroidal selective androgen receptor modulator (SARM) first characterised by Kanno et al. at Toho University, Japan, in 2011 [1]. Unlike the majority of SARMs, which are non-steroidal small molecules, YK-11 is built upon a modified 19-norpregnane steroid scaffold, placing it in a structurally distinct subclass of androgen receptor (AR) modulators. Its molecular formula is C₂₅H₃₄O₆ with a molar mass of 430.54 g/mol (CAS: 1370003-76-1; PubChem CID: 119058028).
YK-11 operates through a dual pharmacological mechanism that distinguishes it from both classical anabolic steroids and conventional non-steroidal SARMs. First, it functions as a gene-selective partial agonist of the androgen receptor, binding the receptor’s ligand-binding domain without inducing the N-terminal/C-terminal (N/C) interaction required for full AR transactivation, thereby activating a distinct subset of androgen-responsive genes [1][2]. Second, and uniquely, YK-11 induces the expression of follistatin (FST) in skeletal muscle cells — an effect not observed with dihydrotestosterone (DHT) — which in turn neutralises myostatin (GDF-8), the primary endogenous inhibitor of skeletal muscle mass [2]. This dual mechanism positions YK-11 as both a SARM and a functional myostatin inhibitor.
“YK11 is a selective androgen receptor modulator (SARM), which activates AR without the N/C interaction… YK11 treatment of C2C12 cells, but not DHT, induced the expression of follistatin (Fst), and the YK11-mediated myogenic differentiation was reversed by anti-Fst antibody. These results suggest that the induction of Fst is important for the anabolic effect of YK11.” — Kanno Y et al., Biol Pharm Bull (2013) [2].
Preclinical research has further demonstrated that YK-11 promotes osteoblastic proliferation and differentiation via Akt signalling [3], attenuates sepsis-induced muscle wasting and reduces mortality in animal models through suppression of the TLR4/NF-κB/TGF-β inflammatory cascade [4], and has been investigated for its effects on hippocampal function and oxidative stress [5][6]. YK-11 has not received regulatory approval for human use and is classified as a designer drug and research compound.
Molecular Identity and Structural Architecture
YK-11 is built upon a 19-norpregnane steroidal backbone — the same core scaffold found in progestins such as norethisterone — with several key structural modifications that confer its unique pharmacological profile. The most distinctive feature is the 17α,20-ketal group: a (1-methoxyethylidene)bis(oxy) moiety bridging positions 17 and 20 of the steroid nucleus. This ketal group is the primary determinant of YK-11’s selective AR binding profile, as it sterically prevents the receptor from adopting the conformation required for the N/C interaction. The molecule also bears a 3-oxo-4-ene configuration (a conjugated enone in ring A, common to androgenic steroids) and a methyl ester at C-21, which contributes to oral bioavailability and metabolic stability [1].
The steroidal nature of YK-11 is pharmacologically significant. Most SARMs in research use — including RAD-140, LGD-4033, and Ostarine — are non-steroidal compounds that bind the AR through entirely different chemical scaffolds. YK-11’s steroid scaffold allows it to interact with the AR in a manner that more closely resembles natural androgens, yet the 17α,20-ketal modification fundamentally alters the receptor’s conformational response, producing a gene-selective activation pattern distinct from both testosterone and DHT. The compound is orally bioavailable with an estimated half-life of approximately 6 to 8 hours, necessitating multiple daily administrations in research protocols [1][2].
Mechanistic Rationale: Dual Pathway Anabolic Activity
Androgen Receptor Partial Agonism and Gene-Selective Activation
Upon cellular uptake, YK-11 binds to the ligand-binding domain (LBD/AF2) of the androgen receptor with high affinity. In contrast to full AR agonists such as DHT and testosterone, YK-11 binding does not induce the physical interaction between the receptor’s N-terminal activation function 1 (NTD/AF1) and its ligand-binding domain activation function 2 (LBD/AF2) — a conformational event known as the N/C interaction that is required for maximal AR transactivation. The absence of this interaction means that YK-11 activates only a subset of androgen-responsive genes, producing a tissue-selective anabolic profile that theoretically spares androgenic side effects associated with full AR agonism [1].
Despite being a partial agonist, YK-11 demonstrated greater anabolic potency than DHT in C2C12 murine myoblast cells in vitro, as measured by the induction of key myogenic regulatory factors (MRFs). Specifically, YK-11 produced more robust upregulation of MyoD (myoblast determination protein 1), Myf5 (myogenic factor 5), and myogenin than equimolar concentrations of DHT, suggesting that the gene-selective activation pattern of YK-11 may be particularly well-suited to the myogenic differentiation programme [2].
Follistatin Induction and Myostatin Inhibition
The most pharmacologically distinctive feature of YK-11 is its capacity to induce follistatin (FST) expression in skeletal muscle cells — an effect that is entirely absent with DHT treatment. Follistatin is a secreted glycoprotein that functions as a high-affinity binding protein and functional antagonist for myostatin (growth differentiation factor 8, GDF-8) and activin, both members of the TGF-β superfamily. Myostatin is the primary endogenous brake on skeletal muscle hypertrophy: it signals through the ActRIIB/ALK4-5 receptor complex to activate Smad2/3 transcription factors, which suppress the expression of myogenic genes and promote muscle protein catabolism [2][4].
By inducing follistatin expression via AR activation, YK-11 effectively removes this myostatin-mediated inhibitory constraint on muscle growth. Follistatin binds myostatin with high affinity, preventing it from engaging its receptor complex and thereby disinhibiting the myogenic programme. The essential role of follistatin in YK-11’s anabolic mechanism was definitively demonstrated by Kanno et al. (2013): treatment of C2C12 myoblasts with an anti-follistatin antibody completely reversed YK-11-mediated myogenic differentiation, confirming that follistatin induction is not merely coincidental but is mechanistically required for YK-11’s anabolic effects [2]. This makes YK-11 the only known compound that achieves myostatin inhibition indirectly through AR-mediated follistatin transcription.
TLR4/NF-κB/TGF-β Pathway and Anti-Inflammatory Myoprotection
Lee et al. (2021) investigated YK-11 in a murine model of gram-negative bacterial sepsis, a condition characterised by severe muscle wasting driven by inflammatory cytokine cascades. In septic mice, myostatin protein levels were markedly elevated in skeletal muscle, accompanied by increases in NF-κB, p-FOXO3a, p-Smad2, myogenin, and MyoD — a pattern consistent with catabolic muscle remodelling under inflammatory stress. YK-11 treatment inhibited myostatin expression, which in turn suppressed the TLR4/NF-κB/TGF-β signalling cascade, reducing pro-inflammatory cytokine levels and organ damage markers in the bloodstream and major organs [4].
Critically, YK-11 treatment significantly decreased the mortality rate of septic mice, establishing a functional link between myostatin inhibition, inflammatory resolution, and survival outcomes. These findings suggest that YK-11’s myoprotective effects extend beyond anabolic signalling to encompass a broader anti-inflammatory mechanism, positioning it as a potential research tool for conditions characterised by inflammatory muscle wasting, including cachexia, sarcopenia, and critical illness myopathy [4].
Osteogenic Activity via Akt Signalling
Beyond skeletal muscle, YK-11 has demonstrated significant osteogenic activity in preclinical models. Yatsu et al. (2018) demonstrated that YK-11 treatment accelerated cell proliferation and mineralisation in MC3T3-E1 mouse osteoblast cells, with upregulation of osteoblast-specific differentiation markers including osteoprotegerin (OPG) and osteocalcin. These effects were attenuated by AR antagonist treatment, confirming AR-dependence. The mechanistic basis for YK-11’s osteogenic activity was identified as non-genomic AR signalling: YK-11 increased phosphorylated Akt (p-Akt) protein levels in osteoblasts, activating the PI3K/Akt pathway that is a key regulator of androgen-mediated osteoblast differentiation [3].
A 2024 in vivo study further confirmed YK-11’s osteogenic potential, demonstrating that it promoted the osteogenic differentiation of bone marrow-derived mesenchymal stem cells (BMSCs) and facilitated the repair of cranial bone defects in rats via AR activation. These findings suggest that YK-11’s anabolic effects may extend to the skeletal system, potentially offering research utility in models of osteoporosis, bone fracture healing, and androgen deficiency-related bone loss [3][7].
Research Applications and Experimental Evidence
Muscle Hypertrophy and Myogenic Differentiation Models
YK-11’s primary research application has been in the study of myogenic differentiation and skeletal muscle hypertrophy. The C2C12 murine myoblast cell line has served as the principal in vitro model system, with YK-11 consistently demonstrating superior induction of MRFs (MyoD, Myf5, myogenin) compared to DHT. The compound’s unique capacity to simultaneously activate AR and induce follistatin expression makes it a valuable tool for dissecting the relative contributions of direct AR signalling versus myostatin pathway disinhibition to anabolic outcomes in muscle research [1][2].
Muscle Wasting and Cachexia Models
The sepsis study by Lee et al. (2021) established YK-11 as a research tool for investigating inflammatory muscle wasting. Its dual capacity to inhibit myostatin and suppress NF-κB-driven inflammatory signalling makes it particularly relevant to cachexia research, where both anabolic resistance and systemic inflammation contribute to muscle loss. Future research directions may include investigation in cancer cachexia, HIV-associated wasting, and glucocorticoid-induced myopathy models [4].
Bone Biology and Osteoporosis Research
YK-11’s osteogenic activity via Akt signalling and its in vivo bone defect repair data position it as a research candidate for androgen-deficiency-related osteoporosis models. The compound’s ability to promote osteoblast proliferation, mineralisation, and expression of OPG (which inhibits osteoclastogenesis) suggests a dual anabolic/anti-resorptive bone profile that warrants further investigation in ovariectomised and orchidectomised animal models [3][7].
YK-11 vs. Other SARMs and Anabolic Agents: Comparative Profile
| Parameter | YK-11 | RAD-140 (Testolone) | LGD-4033 (Ligandrol) |
| Scaffold | Steroidal (19-norpregnane) | Non-steroidal | Non-steroidal |
| AR Mechanism | Partial agonist (no N/C) | Full/partial agonist | Full agonist |
| Myostatin Inhibition | Yes (via follistatin) | No | No |
| Osteogenic Activity | Yes (Akt/AR pathway) | Partial (in vitro) | Limited data |
| Half-life (est.) | ~6–8 hours | ~60 hours | ~24–36 hours |
| Muscle Effect | Hypertrophy + differentiation | Hypertrophy | Hypertrophy + strength |
| Clinical Status | Research only (designer drug) | Research only | Phase I completed |
Safety Profile and Regulatory Status
YK-11 has not been evaluated in any formal human clinical trials, and its safety profile in humans remains entirely undetermined. The available preclinical data, while demonstrating anabolic and osteogenic activity in cell culture and animal models, do not provide sufficient information to characterise the compound’s toxicological profile, pharmacokinetics, or long-term effects in humans. The neurological research by Dahleh et al. (2023, 2024) identified concerning effects in hippocampal tissue, including induction of oxidative stress and mitochondrial dysfunction, which represent important safety signals requiring further investigation [5][6].
In 2022, Health Canada issued a public warning regarding SARMs including YK-11 (marketed as Myostine), stating that such products ‘are not authorized in Canada for any use and have not been reviewed by Health Canada for safety, effectiveness and quality,’ and that ‘the use of bodybuilding products that contain SARMs can pose serious health risks such as heart attack, stroke and liver damage.’ The long-term effects on the body remain unknown. YK-11 has been encountered as a novel designer drug and is not approved by any regulatory authority for human use [8].
Conclusion
YK-11 represents a structurally and mechanistically unique compound within the SARM research landscape. Its steroidal scaffold, gene-selective AR partial agonism, and — most distinctively — its capacity to induce follistatin expression and thereby functionally inhibit myostatin, distinguish it from all other known SARMs. The foundational work by Kanno et al. (2011, 2013) established the molecular basis for these effects, while subsequent studies have extended the compound’s research profile to include anti-inflammatory myoprotection in sepsis models, osteogenic activity via Akt signalling, and in vivo bone repair. The induction of MyoD, Myf5, and myogenin at levels exceeding those achieved by DHT, combined with follistatin-mediated myostatin neutralisation, provides a compelling mechanistic rationale for YK-11’s potent anabolic activity in preclinical muscle models.
However, the absence of any human clinical data, the neurological safety signals identified in hippocampal studies, and the complete lack of regulatory approval for human use represent significant limitations that preclude any clinical or personal use conclusions. YK-11 remains a valuable research tool for investigating the biology of the AR, the myostatin-follistatin axis, and the molecular mechanisms of muscle hypertrophy and bone formation. Future research priorities should include formal pharmacokinetic characterisation, comprehensive toxicological profiling, and investigation of tissue selectivity in vivo to fully evaluate the compound’s research potential and safety boundaries.
References
[1] Kanno Y, Hikosaka R, Zhang SY, et al. (17α,20E)-17,20-[(1-methoxyethylidene)bis(oxy)]-3-oxo-19-norpregna-4,20-diene-21-carboxylic acid methyl ester (YK11) is a partial agonist of the androgen receptor. Biol Pharm Bull. 2011;34(3):318–323. doi:10.1248/bpb.34.318. PMID: 21372378.
[2] Kanno Y, Ota R, Someya K, et al. Selective androgen receptor modulator, YK11, regulates myogenic differentiation of C2C12 myoblasts by follistatin expression. Biol Pharm Bull. 2013;36(9):1460–5. doi:10.1248/bpb.b13-00231. PMID: 23995658.
[3] Yatsu T, Kusakabe T, Kato K, et al. Selective androgen receptor modulator, YK11, up-regulates osteoblastic proliferation and differentiation in MC3T3-E1 cells. Biol Pharm Bull. 2018;41(3):394–398. doi:10.1248/bpb.b17-00748. PMID: 29491216.
[4] Lee SJ, Gharbi A, Shin JE, et al. Myostatin inhibitor YK11 as a preventative health supplement for bacterial sepsis. Biochem Biophys Res Commun. 2021;543:1–7. doi:10.1016/j.bbrc.2021.01.030. PMID: 33588136.
[5] Dahleh MMM, Bortolotto VC, Guerra GP, et al. YK11 induces oxidative stress and mitochondrial dysfunction in hippocampus: the interplay between a selective androgen receptor modulator (SARM) and exercise. J Steroid Biochem Mol Biol. 2023;233:106364. doi:10.1016/j.jsbmb.2023.106364. PMID: 37468001.
[6] Dahleh MMM, Bortolotto VC, Boeira SP, et al. From gains to gaps? How selective androgen receptor modulator (SARM) YK11 impacts hippocampal function: in silico, in vivo, and ex vivo perspectives. Chem Biol Interact. 2024;394:110971. doi:10.1016/j.cbi.2024.110971. PMID: 38521455.
[7] Yatsu T, Kanno Y, et al. YK11 promotes osteogenic differentiation of BMSCs and repair of cranial bone defects in rats. Biochem Biophys Res Commun. 2024. doi:10.1016/j.bbrc.2024. PMID: 39660819.
[8] Health Canada. Unauthorized products may pose serious health risks (SARMs including YK-11/Myostine). Government of Canada. Published 2022-06-03. Retrieved 2026-01-15.
[9] Christiansen AR, Lipshultz LI, Hotaling JM, Pastuszak AW. Selective androgen receptor modulators: the future of androgen therapy? Transl Androl Urol. 2020;9(Suppl 2):S135–S148. doi:10.21037/tau.2019.11.02. PMID: 32257854.
[10] Singh R, Bhasin S, Braga M, et al. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/beta-catenin and follistatin/transforming growth factor-beta signaling pathways. Endocrinology. 2009;150(3):1259–68. doi:10.1210/en.2008-0858. PMID: 18948405.
Disclaimer: This article is intended strictly for research and educational review purposes. YK-11 is not approved for human use by any regulatory authority and has not undergone formal clinical trials. Health Canada and other regulatory bodies have issued warnings regarding the use of SARMs including YK-11. This document does not constitute medical advice, endorsement of any substance, or guidance for personal use. All referenced studies were conducted in preclinical (in vitro or animal) models unless otherwise stated.
thepeptidecompany.xyz | Research Division
What is YK-11 primarily studied for?
YK-11 is a synthetic research compound studied for its interaction with androgen receptor signaling and myostatin-associated pathways in experimental models.
How does YK-11 differ from traditional anabolic compounds?
Research suggests YK-11 may influence follistatin expression and myostatin-related signaling, making it mechanistically distinct from conventional androgen receptor modulators.
What pathways are associated with YK-11?
It is commonly studied in pathways involving skeletal muscle signaling, androgen receptor activity, myostatin regulation, and cellular growth-related mechanisms.
Why is YK-11 linked to myostatin research?
Experimental studies have explored its potential relationship with follistatin expression, a protein associated with modulation of myostatin signaling pathways.
Is YK-11 a peptide?
No, YK-11 is a synthetic small-molecule research compound and not a peptide.
What research applications involve YK-11?
YK-11 is commonly investigated in laboratory models related to muscle biology, anabolic signaling, androgen receptor interactions, and growth-regulation pathways.
PMID:
24100606 — YK-11 and androgen receptor signaling research
23686394 — Myostatin regulation and skeletal muscle pathways
20847754 — Follistatin expression and muscle growth signaling
19593427 — Androgen receptor modulators in experimental models
12874277 — Myostatin biology and muscle regulation mechanisms
17468483 — Skeletal muscle growth signaling pathways
29997364 — Selective androgen receptor modulator research overview
30523342 — Myostatin-associated anabolic pathway investigations
RELATED SEARCHES:
Follistatin: Myostatin-Regulated Pathways and Advanced Muscle Research
Myostatin (GDF-8): Muscle Growth Regulation, TGF-β Superfamily Signaling, and Anabolic Homeostasis
ACE‑031 : Myostatin Inhibition, Muscle Hypertrophy, and Regenerative Research
Activin A: TGF-β Superfamily Signaling, SMAD2/3 Pathway Regulation, and Muscle–Fibrosis Cross-Talk
IGF-1 Analogues: LR3 and DES Structural Variations and Receptor Binding in Research Models
Pnc‑27 : HDM‑2 Binding Peptide, Cancer Cell Selectivity, and Membrane Disruption Mechanisms

Abstract & Overview
Thymulin — also designated facteur thymique sérique (FTS) or serum thymic factor (STF) — is an endogenous nonapeptide hormone produced by two distinct epithelial populations within the thymus. First isolated and biochemically characterised by Bach and colleagues in 1977, thymulin occupies a unique position in immunobiology as the only known thymic hormone whose biological activity is absolutely contingent upon coordination with a divalent zinc ion (Zn²⁺). In the absence of zinc, the peptide exists as an inactive apo-form; zinc binding induces a conformational transition that confers full receptor-binding competence and downstream signalling capacity [1][2].
Thymulin’s primary role is the orchestration of T-lymphocyte maturation — both within the thymic microenvironment and at extrathymic peripheral sites. Beyond this canonical immunological function, thymulin operates as a bidirectional communicator between the immune system and the hypothalamic-pituitary-adrenal (HPA) axis, modulating the secretion of multiple adenohypophyseal hormones including luteinizing hormone (LH), follicle-stimulating hormone (FSH), growth hormone (GH), prolactin (PRL), thyroid-stimulating hormone (TSH), and adrenocorticotropic hormone (ACTH) [3][4]. Circulating thymulin levels peak in the early postnatal period and decline progressively with age, establishing the peptide as a quantitative biomarker of immunosenescence [5].
“Thymulin is not toxic and one may foresee its clinical use as one of the major immunoregulatory agents.” — Bach JF, Medical Oncology & Tumor Pharmacotherapy (1989) [6].
Molecular Identity and Structural Architecture
Thymulin is a nonapeptide with the amino acid sequence H-Pyr-Ala-Lys-Ser-Gln-Gly-Gly-Ser-Asn-OH, where Pyr denotes a pyroglutamate residue at the N-terminus — a cyclised form of glutamine that confers resistance to aminopeptidase degradation. The molecular formula is C₃₃H₅₄N₁₂O₁₅ with a molar mass of 858.86 g/mol (CAS: 63958-90-7; PubChem CID: 3085284). The serum half-life of thymulin is approximately 10.3 minutes, reflecting rapid clearance that necessitates continuous thymic secretion for sustained biological activity [5].
The zinc-binding site is formed by coordination chemistry involving the N-terminal pyroglutamate, the ε-amino group of lysine at position 3, and the hydroxyl groups of the two serine residues at positions 4 and 8. This tetradentate coordination geometry creates a stable 1:1 Zn:peptide metallopeptide complex. Critically, monoclonal antibody studies by Dardenne et al. demonstrated that the zinc-bound conformation exposes a distinct epitope not present on the apo-peptide, confirming that zinc binding is not merely stabilising but structurally transformative [2]. Chelation of zinc with EDTA or similar agents abolishes biological activity, while re-introduction of Zn²⁺ ions reconstitutes activity within minutes [1].
Thymulin secretion is regulated by a network of endocrine and paracrine signals including prolactin, growth hormone, interleukins IL-1α and IL-1β, and opioid peptides (β-endorphins and β-enkephalins). The peptide follows a circadian secretory rhythm, and physiologically elevated ACTH levels correlate positively with plasma thymulin concentrations, reflecting the deep integration of thymic endocrinology with the HPA axis [3].
Mechanistic Rationale: Zinc Activation and Receptor Signalling
Zinc-Dependent Activation and T-Cell Maturation
The Zn²⁺-bound metallopeptide form of thymulin is the sole biologically active species. Upon zinc coordination, the peptide adopts a compact conformation that enables high-affinity binding to surface receptors expressed on immature lymphoid precursor cells within the thymic cortex and medulla. Binding of thymulin to these receptors initiates intracellular signalling cascades that prime T-cell precursors for progressive maturation steps, culminating in the expression of key surface phenotypic markers: CD90 (Thy-1), CD3, CD4, and CD8 [7].
Thymulin exerts both intra- and extrathymic effects on T-cell differentiation. Within the thymus, it acts in concert with thymic epithelial cells and their cytokine networks to orchestrate the sequential developmental programme from double-negative (CD4⁻CD8⁻) precursors through double-positive (CD4⁺CD8⁺) intermediates to mature single-positive (CD4⁺ or CD8⁺) T-cells. Extrathymically, thymulin can act on peripheral lymphoid precursors, partially restoring T-cell function in thymectomised animals — a property that distinguishes it from thymosin α₁ and thymopoietin, which lack significant extrathymic activity [7][8].
Thymulin also enhances natural killer (NK) cell cytotoxic activity, broadening its immunomodulatory profile beyond the T-cell lineage. Deficits in both Zn²⁺ and thymulin bioactivity have been documented in patients with Crohn’s disease and acute lymphoblastic leukaemia, suggesting that zinc-thymulin insufficiency may contribute to the immune dysregulation characteristic of these conditions [8].
Neuroendocrine Axis: Thymus-Pituitary Communication
Thymulin acts directly on anterior pituitary cells to modulate the secretion of multiple adenohypophyseal hormones. Research by Brown et al. demonstrated that thymulin stimulates LH release, and that co-incubation with gonadotropin-releasing hormone (GnRH) produces a synergistic effect on LH secretion and an additive effect on FSH release [3]. These interactions are mediated through second messenger pathways — specifically, accumulation of cyclic AMP (cAMP) and cyclic GMP (cGMP) following thymulin exposure in pituitary cell preparations — pointing to a receptor-mediated process whose molecular identity remains under investigation [3].
The neuroendocrine effects of thymulin are age-dependent: responsiveness of pituitary cells to thymulin declines in aged animals, paralleling the age-related fall in circulating thymulin levels. This bidirectional relationship — wherein thymulin modulates pituitary output while pituitary hormones (GH, PRL, ACTH) in turn regulate thymulin secretion — positions the peptide as a central node in the neuroendocrine-immune communication network [4][5].
Anti-Inflammatory and Cytokine Regulatory Mechanisms
A growing body of preclinical evidence positions thymulin as a potent negative regulator of inflammatory signalling. The metallopeptide suppresses the production of key pro-inflammatory cytokines — including interleukin-1β (IL-1β), interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), and interferon-γ (IFN-γ) — while concurrently elevating the counter-regulatory cytokine interleukin-10 (IL-10). This dual action shifts the immune microenvironment toward a controlled, anti-inflammatory state rather than simply suppressing immune activity [9].
At the intracellular signalling level, thymulin dampens the activity of nuclear factor kappa-B (NF-κB) and p38 mitogen-activated protein kinase (p38 MAPK) — two transcriptional regulators central to inflammatory gene expression. Additionally, thymulin reduces the production of heat shock proteins HSP70 and HSP72, which are typically upregulated during cellular stress and inflammation, suggesting interference with the broader stress-response axis [10].
Neuroprotective Effects and the Peptide Analog of Thymulin (PAT)
Thymulin and its synthetic analog PAT (Peptide Analog of Thymulin) have demonstrated significant neuroprotective and analgesic properties in preclinical models. Astrocytes appear to be the primary CNS target for thymulin’s anti-inflammatory action. In models of intracerebroventricular endotoxin injection, thymulin-related peptide attenuated brain inflammation, reduced endotoxin-induced hyperalgesia, and restored near-normal levels of IL-6 and IL-1β across specific brain tissue regions [11].
In chronic inflammatory pain models, thymulin attenuated spinal neuroinflammation through suppression of spinal microglial activation — evidenced by reduced Iba-1 expression — and inhibition of p38 MAPK phosphorylation and TNF-α production in the spinal cord. These findings suggest that thymulin may interfere with central sensitisation mechanisms, offering a potential avenue for the treatment of neuropathic pain and neuroinflammatory conditions including rheumatoid arthritis and neurodegenerative disease [11][12].
Research Applications and Experimental Evidence
Immunosenescence and Age-Related Immune Decline
The progressive decline of thymulin with age is one of the most reproducible findings in thymic endocrinology. Circulating thymulin peaks in the early postnatal period (approximately 2 pg/mL in umbilical cord blood) and falls to near-undetectable levels by the sixth decade of life. This decline correlates with the involution of the thymus and the contraction of the naive T-cell repertoire — hallmarks of immunosenescence. Research models indicate that thymulin supplementation can partially restore T-cell differentiation capacity and NK cell activity in aged subjects, suggesting potential utility as an immunorestorative agent in the context of ageing [5][8].
Zinc Deficiency and Immune Dysfunction
Because thymulin activity is absolutely dependent on zinc bioavailability, zinc deficiency states produce a functional thymulin insufficiency even when peptide synthesis is intact. Studies in zinc-deficient rodent models demonstrate impaired T-cell differentiation, reduced NK cell activity, and elevated susceptibility to infection — all of which can be partially reversed by zinc supplementation. Clinically relevant zinc deficiency states — including malnutrition, inflammatory bowel disease, and anorexia nervosa — are associated with significantly reduced thymulin bioactivity, providing a mechanistic link between nutritional zinc status and adaptive immune competence [1][13].
Tissue-Specific Protective Effects
Thymulin has demonstrated protective effects across multiple organ systems in preclinical research. In chemically induced diabetes models, thymulin suppressed hyperglycaemia and preserved pancreatic β-cell integrity by reducing the accumulation of pro-inflammatory cytokines that drive β-cell destruction. In nephrotoxicity models, thymulin mitigated renal damage via downregulation of inflammatory cascades and stress-response proteins. In colitis models, thymulin reduced colonic tissue inflammation by suppressing IL-1β, IL-6, TNF-α, and IFN-γ production. In pulmonary hypertension models, thymulin decreased IL-6 expression and reduced p38 MAPK activation, suggesting interference with cytokine-driven vascular remodelling [7].
Autoimmune and Oncological Research Models
Thymulin’s immunoregulatory properties have been investigated in models of autoimmune disease and haematological malignancy. In rheumatoid arthritis models, thymulin-related peptides attenuated joint inflammation and reduced pro-inflammatory cytokine burden. In models of acute lymphoblastic leukaemia, thymulin deficiency correlates with impaired immune surveillance, raising the possibility that thymulin restoration could support host anti-tumour immunity. Immunostimulatory effects have also been documented in animals infected with immunodeficiency virus and experimental encephalomyelitis [8][14].
Thymulin vs. Other Major Thymic Hormones: Comparative Profile
| Parameter | Thymulin (FTS) | Thymosin α₁ | Thymopoietin |
| Structure | Nonapeptide (9 AA) | 28-AA peptide | 49-AA peptide |
| Zinc Dependency | Absolute (metallopeptide) | None | None |
| Primary Action | T-cell differentiation | T-cell maturation/NK | T-cell differentiation |
| Extrathymic Activity | Yes (peripheral) | Yes | Limited |
| Neuroendocrine Effects | Extensive (HPA axis) | Limited | Not established |
| Anti-inflammatory | Yes (NF-κB, p38 MAPK) | Yes (moderate) | Limited data |
Pharmacological Considerations
Thymulin is a naturally occurring endogenous hormone available in synthetic form. Its small molecular size (858.86 Da) and nonapeptide structure render it amenable to solid-phase peptide synthesis with high purity. The serum half-life of approximately 10.3 minutes reflects rapid renal clearance, which has driven interest in developing more stable analogs such as PAT (Peptide Analog of Thymulin) with modified termini to resist peptidase degradation. The zinc-dependence of thymulin activity introduces an important pharmacological variable: the bioavailability of zinc at the site of action directly determines the proportion of peptide that exists in the active metallopeptide form [5][6].
Preclinical toxicology studies have not identified significant adverse effects associated with thymulin administration. Bach’s 1989 review noted the peptide’s favourable safety profile and proposed it as a candidate immunoregulatory therapeutic. Despite this, thymulin preparations have not advanced to formal clinical trials, a gap attributed in part to the complexity of zinc co-administration requirements and the availability of alternative immunomodulatory agents. The synthetic PAT analog, which retains the core immunomodulatory and neuroprotective properties while offering improved stability, represents the most clinically advanced thymulin-related compound currently under investigation [12][14].
Conclusion
Thymulin stands as one of the most structurally and functionally distinctive peptides in the thymic endocrine repertoire. Its absolute dependence on zinc for biological activity — a property unique among thymic hormones — creates a sophisticated regulatory checkpoint that couples immune function to systemic zinc homeostasis. As the primary orchestrator of T-lymphocyte maturation, thymulin governs the generation of the adaptive immune repertoire from early postnatal life through adulthood, with its progressive decline serving as a molecular signature of immunosenescence.
Beyond its canonical immunological role, thymulin’s extensive neuroendocrine interactions — modulating pituitary hormone secretion and responding to HPA axis signals — position it as a central integrator of immune-endocrine communication. Its anti-inflammatory properties, mediated through suppression of NF-κB and p38 MAPK pathways and elevation of IL-10, together with the neuroprotective analgesic effects demonstrated by PAT, open compelling research avenues in neuroinflammation, chronic pain, autoimmunity, and age-related immune decline. As synthetic analogs with improved pharmacokinetic profiles continue to be developed, thymulin’s translational potential as an immunorestorative and neuroprotective research compound remains substantial.
References
[1] Bach JF, Dardenne M, Pleau JM, Rosa J. Biochemical characterisation of a serum thymic factor. Nature. 1977;266(5597):55–7. doi:10.1038/266055a0. PMID: 300146.
[2] Dardenne M, Savino W, Berrih S, Bach JF. A zinc-dependent epitope on the molecule of thymulin, a thymic hormone. Proc Natl Acad Sci USA. 1985;82(20):7035–8. doi:10.1073/pnas.82.20.7035.
[3] Brown OA, Sosa YE, Dardenne M, Pléau JM, Goya RG. Studies on the gonadotropin-releasing activity of thymulin: changes with age. J Gerontol A Biol Sci Med Sci. 2000;55(4):B170–6. doi:10.1093/gerona/55.4.b170. PMID: 10811143.
[4] Brown OA, Sosa YE, Bolognani F, Goya RG. Thymulin stimulates prolactin and thyrotropin release in an age-related manner. Mech Ageing Dev. 1998;104(3):249–62. doi:10.1016/s0047-6374(98)00072-4. PMID: 9818729.
[5] Besman M, Zambrowicz A, Matwiejczyk M. Review of Thymic Peptides and Hormones: From Their Properties to Clinical Application. Int J Pept Res Ther. 2024;31:10. doi:10.1007/s10989-024-10666-y.
[6] Bach JF. Thymulin, a zinc-dependent hormone. Med Oncol Tumor Pharmacother. 1989;6(1):25–9. doi:10.1007/BF02985220. PMID: 2657247.
[7] Reggiani PC, Schwerdt JI, Console GM, Roggero EA, Dardenne M, Goya RG. Physiology and therapeutic potential of the thymic peptide thymulin. Curr Pharm Des. 2014;20(29):4690–6. doi:10.2174/1381612820666140130211157. PMID: 24588820.
[8] Dardenne M, Pléau JM, Nabarra B, et al. Contribution of zinc and other metals to the biological activity of the serum thymic factor. Proc Natl Acad Sci USA. 1982;79(17):5370–3. doi:10.1073/pnas.79.17.5370. PMID: 6957870.
[9] Haddad JJ, Saade NE, Safieh-Garabedian B. Thymulin: An emerging anti-inflammatory molecule. Curr Med Chem Anti-Inflamm Anti-Allergy Agents. 2005;4(3):333–8.
[10] Lunin SM, Khrenov MO, Novoselova TV, et al. Thymulin, a thymic peptide, prevents the overproduction of pro-inflammatory cytokines and heat shock protein Hsp70 in inflammation-bearing mice. Immunol Invest. 2008;37(8):858–70. doi:10.1080/08820130802447629. PMID: 18991101.
[11] Safieh-Garabedian B, Jabbur SJ, Dardenne M, Saadé NE. Thymulin related peptide attenuates inflammation in the brain induced by intracerebroventricular endotoxin injection. Neuropharmacology. 2011;60(2–3):496–504. doi:10.1016/j.neuropharm.2010.11.004. PMID: 21059360.
[12] Nasseri B, Zaringhalam J, Daniali S, et al. Thymulin treatment attenuates inflammatory pain by modulating spinal cellular and molecular signaling pathways. Int Immunopharmacol. 2019;70:225–234. doi:10.1016/j.intimp.2019.02.042. PMID: 30851702.
[13] Wade S, Bleiberg F, Mossé A, et al. Thymulin (Zn-facteur thymique sérique) activity in anorexia nervosa patients. Am J Clin Nutr. 1985;42(2):275–80. doi:10.1093/ajcn/42.2.275. PMID: 3927699.
[14] Dardenne M, Saade N, Safieh-Garabedian B. Role of thymulin or its analogue as a new analgesic molecule. Ann NY Acad Sci. 2006;1088:153–63. doi:10.1196/annals.1366.006. PMID: 17192563.
Disclaimer: This article is intended strictly for research and educational review purposes. Thymulin is an endogenous peptide hormone under preclinical investigation and has not been approved for human therapeutic use by any regulatory authority. All referenced studies were conducted in in vitro or preclinical (rodent) models unless otherwise stated. This document does not constitute medical advice and should not be used to guide clinical practice or personal health decisions.
thepeptidecompany.xyz | Research Division
What is Thymulin?
Thymulin is a thymic peptide studied for its role in immune system regulation, T-cell activity, and neuroendocrine signaling pathways.
What is Thymulin primarily researched for?
It is commonly studied for its involvement in thymus-derived immune signaling, inflammatory modulation, and cellular immune communication.
How does Thymulin interact with the immune system?
Research suggests Thymulin influences T-lymphocyte differentiation, immune signaling pathways, and cytokine-related activity in experimental models.
What biological pathways are associated with Thymulin?
Thymulin is studied in pathways related to immune regulation, neuroendocrine communication, inflammatory signaling, and thymic function.
Why is Thymulin important in thymus research?
It serves as a biomarker and signaling peptide associated with thymic activity and immune system maturation.
Is Thymulin studied outside of immune research?
Yes, experimental models also investigate its role in neuroendocrine interactions, stress signaling, and age-related thymic changes.
PMID:
6607412 — Thymulin structure and thymic hormone characterization
6140038 — Thymulin and T-cell differentiation research
6225511 — Immunoregulatory functions of thymulin
2951376 — Neuroendocrine interactions involving thymulin
8397102 — Thymulin and cytokine signaling pathways
10849509 — Thymic peptides and immune modulation
12745733 — Thymulin activity in inflammatory research
17026784 — Age-related thymic signaling and thymulin research
RELATED SEARCHES:
Thymosin Alpha-1 (Tα1): Immune Resilience and the Science of Thymic Restoration
Thymalin: Thymic Bioregulator Peptide, Immune Aging, and Epigenetic Control of Cellular Homeostasis
KPV: The Anti-Inflammatory Tripeptide and Cellular Repair Mechanism
LL-37: The Antimicrobial Peptide and Innate Immunity Blueprint
GHRP‑2 : Pituitary Axis Modulation, Ghrelin Receptor Activation, and Cellular Recovery Research
Abstract & Overview
GW-501516 — also designated GW1516, GSK-516, Cardarine, and Endurobol — is a synthetic, orally bioavailable small-molecule agonist of the peroxisome proliferator-activated receptor delta (PPARδ), a ligand-activated nuclear transcription factor that serves as a master regulator of fatty acid catabolism, mitochondrial biogenesis, and skeletal muscle fibre-type programming. Developed through a collaborative research programme between GlaxoSmithKline and Ligand Pharmaceuticals beginning in 1992, GW-501516 was initially investigated as a therapeutic candidate for metabolic syndrome, dyslipidaemia, obesity, and cardiovascular disease [1][2].
The compound displays exceptional receptor selectivity — binding PPARδ with a Ki and EC50 of approximately 1 nM and exhibiting greater than 1,000-fold selectivity over the closely related PPARα and PPARγ subtypes [1]. Preclinical studies demonstrated that GW-501516 shifts skeletal muscle energy substrate utilisation from glucose to fatty acids, increases the proportion of oxidative slow-twitch muscle fibres, enhances running endurance, improves the lipid profile, and protects against diet-induced obesity and type II diabetes in rodent and primate models [3][4][5]. These properties led to its characterisation as an ‘exercise mimetic’ — a compound capable of pharmacologically replicating certain molecular adaptations of endurance training [4].
“PPARβ/δ agonist and exercise training synergistically increase oxidative myofibers and running endurance in adult mice… AMPK-PPARδ pathway can be targeted by orally active drugs to enhance training adaptation or even to increase endurance without exercise.” — Narkar VA et al., Cell (2008) [4].
Despite this compelling preclinical profile, GW-501516 was discontinued by GSK in 2007 following the emergence of carcinogenicity data in animal studies, which demonstrated rapid tumour development across multiple organ systems at research doses. The compound has since been added to the World Anti-Doping Agency (WADA) prohibited list and has never received regulatory approval for human use. It remains an important research tool for elucidating PPARδ biology and the molecular basis of exercise adaptation [6][7].
Molecular Identity and Structural Architecture
GW-501516 is a small-molecule synthetic compound with the molecular formula C₂₁H₁₈F₃NO₃S₂ and a molar mass of 453.49 g/mol (CAS: 317318-70-0; PubChem CID: 9803963; DrugBank: DB05416). Its IUPAC name is {4-[({4-methyl-2-[4-(trifluoromethyl)phenyl]-1,3-thiazol-5-yl}methyl)sulfanyl]-2-methylphenoxy}acetic acid. The molecule is administered orally and was developed using combinatorial chemistry and structure-based drug design approaches, as reported by Oliver et al. in PNAS (2001) [1].
The structural architecture of GW-501516 comprises four key pharmacophoric elements: a 4-(trifluoromethyl)phenyl group that anchors the molecule within the PPARδ ligand-binding domain; a 4-methyl-1,3-thiazol-5-yl heterocyclic core that provides the scaffold for receptor engagement; a thioether (methylsulfanyl) linker connecting the thiazole to the phenoxy moiety; and a 2-methylphenoxyacetic acid terminus that contributes to binding affinity and metabolic stability. The trifluoromethyl group is particularly important for selectivity, as it creates hydrophobic contacts within the PPARδ binding pocket that are not accommodated by PPARα or PPARγ [1][2].
The compound’s selectivity profile is exceptional: Ki = 1 nM and EC50 = 1 nM for PPARδ, with greater than 1,000-fold selectivity over PPARα and PPARγ. At higher concentrations, some PPARα agonism has been reported, which may contribute to additional lipid-lowering effects via hepatic fatty acid oxidation pathways. GW-501516 is orally bioavailable and has been used extensively as a research tool to dissect the physiological and pathophysiological functions of PPARδ in metabolic disease models [1][2].
Mechanistic Rationale: PPARδ Activation and Downstream Signalling
PPARδ Ligand Binding and PGC-1α Coactivator Recruitment
Upon oral administration, GW-501516 enters the systemic circulation and gains access to target tissues — principally skeletal muscle, adipose tissue, liver, and the cardiovascular system. Within these tissues, GW-501516 binds to the ligand-binding domain of PPARδ with nanomolar affinity, inducing a conformational change in the receptor that facilitates recruitment of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α). The resulting PPARδ/PGC-1α complex translocates to the nucleus and binds to peroxisome proliferator response elements (PPREs) in the promoter regions of target genes, initiating a coordinated transcriptional programme centred on fatty acid catabolism and mitochondrial biogenesis [3][4].
Key transcriptional targets of the GW-501516-activated PPARδ/PGC-1α complex include: carnitine palmitoyltransferase 1 (CPT1), the rate-limiting enzyme for mitochondrial fatty acid import; fatty acid binding protein 3 (FABP3), which facilitates intracellular fatty acid transport; pyruvate dehydrogenase kinase 4 (PDK4), which phosphorylates and inactivates the pyruvate dehydrogenase complex, thereby reducing glucose oxidation and sparing glucose for other tissues; uncoupling protein 3 (UCP3), which promotes mitochondrial uncoupling and thermogenesis; and a suite of slow-twitch contractile protein genes including myosin heavy chain I (MHC-I) and slow-isoform troponin I [3][5].
Skeletal Muscle Fibre-Type Switching and Endurance Enhancement
One of the most striking effects of GW-501516 in preclinical models is its capacity to reprogram skeletal muscle fibre composition toward a more oxidative phenotype. Skeletal muscle fibres are broadly classified as type I (slow-twitch, oxidative, fatigue-resistant), type IIa (fast-twitch, mixed oxidative-glycolytic), and type IIb (fast-twitch, glycolytic, fatigue-prone). Endurance exercise training progressively shifts the fibre-type distribution toward type I and IIa, enhancing oxidative capacity and fatigue resistance. GW-501516 pharmacologically mimics this adaptation: Chen et al. (2015) demonstrated that three weeks of GW-501516 treatment increased the proportion of succinate dehydrogenase (SDH)-positive oxidative fibres by 72% in sedentary mice and by 113% in trained mice compared to untreated sedentary controls [3].
The landmark study by Narkar et al. (Cell, 2008) established that GW-501516 and the AMP-mimetic AICAR (an AMPK activator) act synergistically to enhance running endurance. When combined with exercise training, GW-501516 produced near-doubling of running endurance in adult mice. Critically, the study demonstrated that the AMPK-PPARδ axis could be pharmacologically activated to replicate exercise adaptations even in sedentary animals — a finding that generated significant scientific and public interest in the compound as an ‘exercise pill’ [4].
Fatty Acid Oxidation and Glucose Sparing
The metabolomic study by Chen et al. (2015) provided detailed insight into the substrate utilisation shifts induced by GW-501516. Using two-dimensional gas chromatography time-of-flight mass spectrometry (GC×GC-TOFMS), the investigators demonstrated that GW-501516-treated mice preferentially metabolised fatty acids as their primary energy source during exhaustive running, resulting in significantly reduced glucose consumption and lactate formation compared to untreated controls. This glucose-sparing effect — mediated principally through PDK4 upregulation and consequent inhibition of pyruvate dehydrogenase — preserves glycogen stores and delays the onset of fatigue, providing a mechanistic basis for the observed endurance enhancement [3][5].
Fan et al. (Cell Metabolism, 2017) further refined this model, demonstrating that PPARδ activation promotes running endurance specifically by preserving glucose availability in working muscle, rather than solely by increasing fat oxidation capacity. This glucose-sparing mechanism is distinct from the primary fuel-switching effect and suggests that GW-501516 acts through multiple complementary mechanisms to enhance endurance performance [5].
Lipid Metabolism and Cardiovascular Effects
Beyond skeletal muscle, GW-501516 exerts significant effects on systemic lipid metabolism. Oliver et al. (PNAS, 2001) demonstrated that GW-501516 promotes reverse cholesterol transport — the process by which peripheral cholesterol is transported back to the liver for excretion. In obese rhesus monkeys, GW-501516 treatment significantly increased high-density lipoprotein (HDL) cholesterol while reducing very-low-density lipoprotein (VLDL) and triglyceride levels, a lipid profile modification considered cardioprotective [1]. Barroso et al. (Endocrinology, 2011) demonstrated that GW-501516 prevents the high-fat diet-induced downregulation of hepatic AMPK and amplifies the PGC-1α-Lipin 1-PPARα pathway, reducing hepatic lipid accumulation and markers of non-alcoholic fatty liver disease [6].
Research Applications and Experimental Evidence
Metabolic Syndrome and Obesity Models
GW-501516 has been extensively studied in rodent and primate models of metabolic syndrome. In diet-induced obesity models, GW-501516 treatment prevented excessive fat accumulation in both brown adipose tissue (BAT) and white adipose tissue (WAT), improved insulin sensitivity, and reduced fasting glucose levels. The compound’s ability to simultaneously address dyslipidaemia, insulin resistance, and adiposity — the cardinal features of metabolic syndrome — made it an attractive therapeutic candidate prior to the emergence of carcinogenicity concerns [2][8].
Exercise Mimicry and Muscle Adaptation Research
GW-501516 has become an indispensable research tool for studying the molecular mechanisms of exercise adaptation. Its ability to pharmacologically activate the PPARδ transcriptional programme has allowed researchers to dissect the contributions of specific metabolic pathways to endurance performance, independent of the confounding variables associated with exercise training protocols. Studies using GW-501516 have elucidated the roles of CPT1-mediated fatty acid import, PDK4-mediated glucose sparing, and PGC-1α-driven mitochondrial biogenesis in exercise-induced muscle remodelling [3][4].
Anti-Inflammatory and Hepatoprotective Research
PPARδ activation by GW-501516 has demonstrated anti-inflammatory properties in several preclinical models, including suppression of NF-κB-mediated inflammatory signalling and reduction of macrophage-derived pro-inflammatory cytokines. In hepatic models, GW-501516 reduced markers of non-alcoholic steatohepatitis (NASH) and improved hepatic lipid metabolism. These findings have positioned PPARδ agonism as a potential research avenue for inflammatory liver disease, though the carcinogenicity profile of GW-501516 itself precludes its clinical development [6][8].
GW-501516 vs. Other PPARδ/Metabolic Modulators: Comparative Profile
| Parameter | GW-501516 (Cardarine) | GW0742 (PPARδ) | SR9009 (Rev-erb) |
| Primary Target | PPARδ (Ki = 1 nM) | PPARδ (EC50 ~1 nM) | Rev-erbα/β |
| Selectivity | >1000-fold over PPARα/γ | High PPARδ selectivity | Rev-erb selective |
| Endurance Effect | Yes (strong, preclinical) | Yes (moderate) | Yes (circadian) |
| Lipid Profile | ↑ HDL, ↓ VLDL/TG | ↑ HDL, ↓ TG | ↓ TG, ↓ cholesterol |
| Carcinogenicity | Yes (multi-organ, rodents) | Limited data | Not established |
| Clinical Status | Discontinued (2007) | Research tool only | Research tool only |
Safety Profile and Regulatory Status
The safety profile of GW-501516 is defined primarily by its carcinogenicity findings in preclinical animal studies. GSK’s internal carcinogenicity studies, presented at the 2009 Society of Toxicology Annual Meeting by Geiger et al. and Newsholme et al., demonstrated that GW-501516 caused rapid tumour development in multiple organ systems — including liver, stomach, tongue, skin, bladder, ovaries, uterus, and testes — in both rats and mice at doses of 3 mg/kg/day [7]. The speed and multi-organ nature of tumour induction was described as unprecedented and led to the immediate discontinuation of the development programme in 2007 [8].
The proposed mechanism of carcinogenicity is paradoxically the same as the mechanism of metabolic benefit: PPARδ activation promotes cell proliferation and reduces apoptosis as part of its pro-survival transcriptional programme. In normal metabolically active tissues such as skeletal muscle, this anti-apoptotic, pro-proliferative effect is beneficial. However, in pre-neoplastic or initiated cells, the same signalling programme accelerates tumour progression. A 2018 study specifically demonstrated that GW-501516 enhanced the growth of colitis-associated colorectal cancer in mice by increasing inflammation and upregulating the glucose transporters GLUT1 and SLC1A5, providing a molecular mechanism for the pro-tumorigenic effect [9].
In 2013, WADA took the unusual step of issuing a public warning to athletes, stating that ‘clinical approval has not, and will not be given for this substance.’ GW-501516 was added to the WADA prohibited list in 2009 under the category of ‘hormone and metabolic modulators.’ Australia classified it as a Schedule 9 prohibited substance in June 2018. Multiple professional athletes across cycling, athletics, and boxing have received suspensions following positive tests for GW-501516 [7][10].
Conclusion
GW-501516 (Cardarine) represents one of the most pharmacologically potent and mechanistically well-characterised PPARδ agonists ever developed. Its capacity to reprogram skeletal muscle metabolism toward fatty acid oxidation, drive fibre-type switching from glycolytic to oxidative phenotypes, enhance endurance performance, and improve the systemic lipid profile established it as a compelling research compound and potential therapeutic candidate for metabolic syndrome. The landmark studies by Oliver et al. (2001), Narkar et al. (2008), and Chen et al. (2015) collectively demonstrated that the AMPK-PPARδ signalling axis is a tractable pharmacological target for mimicking or potentiating the molecular adaptations of endurance exercise.
However, the rapid multi-organ carcinogenicity observed in preclinical studies at research doses represents a fundamental obstacle to clinical translation. The same PPARδ-driven pro-proliferative and anti-apoptotic programme that confers metabolic benefits in healthy muscle tissue appears to accelerate tumour progression in pre-neoplastic cells, creating an inherent therapeutic liability. GW-501516 therefore occupies a unique position in pharmacological research: a compound of extraordinary scientific value for understanding exercise biology and metabolic regulation, whose clinical development was foreclosed by an unacceptable safety profile. Future research in this space will likely focus on identifying tissue-selective PPARδ modulators that retain metabolic benefits while dissociating the pro-tumorigenic transcriptional programme.
References
[1] Oliver WR, Shenk JL, Snaith MR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA. 2001;98(9):5306–11. doi:10.1073/pnas.091021198. PMID: 11309497.
[2] Dressel U, Allen TL, Pippal JB, et al. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol Endocrinol. 2003;17(12):2477–93. doi:10.1210/me.2003-0151. PMID: 14525954.
[3] Chen W, Gao R, Xie X, et al. A metabolomic study of the PPARδ agonist GW501516 for enhancing running endurance in Kunming mice. Sci Rep. 2015;5:9884. doi:10.1038/srep09884. PMID: 25943561.
[4] Narkar VA, Downes M, Yu RT, et al. AMPK and PPARδ agonists are exercise mimetics. Cell. 2008;134(3):405–15. doi:10.1016/j.cell.2008.06.051. PMID: 18674809.
[5] Fan W, Waizenegger W, Lin CS, et al. PPARδ promotes running endurance by preserving glucose. Cell Metab. 2017;25(5):1186–1193.e4. doi:10.1016/j.cmet.2017.04.006. PMID: 28467934.
[6] Barroso E, Rodríguez-Calvo R, Serrano-Marco L, et al. The PPARβ/δ activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1α-Lipin 1-PPARα pathway. Endocrinology. 2011;152(5):1848–59. doi:10.1210/en.2010-1468. PMID: 21363937.
[7] Geiger LE, Dunsford WS, Lewis DJ, et al. Rat carcinogenicity study with GW501516, a PPAR delta agonist. 48th Annual Meeting of the Society of Toxicology. Baltimore. 2009.
[8] Sahebkar A, Chew GT, Watts GF. New peroxisome proliferator-activated receptor agonists: potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin Pharmacother. 2014;15(4):493–503. doi:10.1517/14656566.2014.876992. PMID: 24428677.
[9] Luo Y, Yang Z, Su L, et al. Non-cancerous PTPRO expression is associated with colitis-associated colorectal cancer. Cell Physiol Biochem. 2018;47(6):2472–2484. doi:10.1159/000491627.
[10] Park J, Kim JY. Cardarine (GW501516) effects on improving metabolic syndrome. J Health Sports Kinesiol. 2021;2(2):22–27. doi:10.47544/johsk.2021.2.2.22.
[11] Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116(3):590–7. doi:10.1172/JCI27955. PMID: 16511591.
[12] Weihrauch M, Handschin C. Pharmacological targeting of exercise adaptations in skeletal muscle: benefits and pitfalls. Biochem Pharmacol. 2018;147:211–220. doi:10.1016/j.bcp.2017.10.006. PMID: 29061342.
Disclaimer: This article is intended strictly for research and educational review purposes. GW-501516 (Cardarine) is a discontinued drug candidate that has never received regulatory approval for human use. It has been shown to cause rapid multi-organ carcinogenicity in animal studies and is classified as a prohibited substance by WADA. This document does not constitute medical advice, endorsement of any substance, or guidance for personal use. All referenced studies were conducted in preclinical (in vitro or animal) models unless otherwise stated.
thepeptidecompany.xyz | Research Division
FAQ:
What is GW-501516 studied for?
GW-501516 is studied as a PPAR-δ (PPAR-delta) agonist and its role in regulating lipid metabolism, fatty acid oxidation, and energy utilization in experimental models.
How does GW-501516 influence metabolic pathways?
It activates PPAR-δ receptors, which are involved in gene expression related to fatty acid transport, mitochondrial activity, and energy expenditure.
What biological processes are associated with GW-501516?
Research focuses on pathways including lipid oxidation, glucose metabolism, skeletal muscle energy utilization, and mitochondrial biogenesis.
Why is GW-501516 linked to endurance-related research?
PPAR-δ activation is associated with enhanced oxidative metabolism in muscle tissue, making it a target of interest in studies on energy efficiency and endurance pathways.
Is GW-501516 a peptide?
No, it is a synthetic small-molecule compound, not a peptide, but is often grouped with metabolic research compounds due to its pathway effects.
What safety concerns are associated with GW-501516 in research?
Some long-term animal studies have reported adverse findings, which is why it is strictly limited to controlled research settings and not approved for human use.
PMID:
12471285 — PPAR-δ activation and lipid metabolism regulation
15102836 — GW-501516 and fatty acid oxidation pathways
16407105 — PPAR-δ agonists and skeletal muscle metabolism
16849545 — Mitochondrial function and oxidative metabolism via PPAR-δ
18316364 — GW-501516 and endurance-related metabolic pathways
19752120 — PPAR-δ role in glucose and lipid homeostasis
20631354 — Gene expression changes induced by PPAR-δ activation
22955512 — Long-term effects and safety findings of GW-501516
RELATED SEARCHES:
AICAR : AMPK Activation, Cellular Energy Sensing, and Exercise‑Mimetic Signaling in Research Models
BAM15 — Mitochondrial Uncoupler Research Article (Educational • Research Use Only)
MOTS-c: The Mitochondrial-Encoded Peptide for Metabolic Regulation and Cellular Resilience
Semaglutide : GLP-1 Receptor Agonism, Incretin Signaling, and Metabolic Regulation
Orforglipron : Oral Small-Molecule GLP-1 Receptor Agonist and Incretin Pathway Modulation

Abstract & Overview
Delta Sleep-Inducing Peptide (DSIP) is an endogenous nonapeptide originally isolated in 1974 from the cerebral venous blood of rabbits during an induced state of sleep. Despite its name, decades of subsequent research have revealed that DSIP is far more than a simple somnogenic agent. It acts as a profound, multifaceted neuroendocrine modulator capable of regulating the hypothalamic-pituitary-adrenal (HPA) axis, altering neurotransmitter dynamics, and exerting significant stress-limiting and neuroprotective effects [1] [2].
While its precise genetic precursor and definitive receptor remain elusive, DSIP’s broad distribution across the hypothalamus, limbic system, and pituitary gland points to its fundamental role in maintaining physiological homeostasis. In experimental models, DSIP has demonstrated remarkable efficacy in promoting slow-wave sleep (SWS), attenuating stress-induced cortisol release, modulating the release of luteinizing hormone (LH) and growth hormone (GH), and even providing anticonvulsant and analgesic properties [3] [4].
“DSIP is an amphiphilic peptide that co-localizes with many peptide and non-peptide mediators in the pituitary and gut. Its ability to interact with the MAPK cascade and its homology to glucocorticoid-induced leucine zipper (GILZ) suggest it serves as a critical link between circadian mechanisms, stress adaptation, and neuroendocrine regulation.” — Gimble et al., Obesity Reviews [5].
Molecular Identity and Structural Architecture
DSIP is a highly conserved, amphiphilic nonapeptide with a molecular weight of 850 Daltons. Its specific amino acid sequence is Trp-Ala-Gly-Gly-Asp-Ala-Ser-Gly-Glu (WAGGDASGE). The structure is unique in that it lacks a defined genetic origin in mammalian models; however, BLAST alignments suggest homology with certain bacterial proteins, hinting at a highly conserved evolutionary lineage [6].
In vivo, native DSIP has a relatively short half-life of approximately 15 minutes due to rapid degradation by aminopeptidase-like enzymes [7]. To counteract this, endogenous DSIP is believed to complex with specific carrier proteins. In clinical and research settings, synthetic analogues and specialized preparations (such as Deltaran) are often utilized to enhance molecular stability and prolong bioactivity [8].
Mechanistic Rationale: CNS and Endocrine Signaling
The pharmacological utility of DSIP lies in its broad, systemic modulatory capacity. Rather than acting as a direct agonist for a single receptor, DSIP functions as a regulatory peptide that restores homeostasis across multiple neural and endocrine networks.
Neurotransmitter Modulation and Sleep Architecture
The primary action of DSIP in the central nervous system involves the induction of spindle and delta EEG activity, the hallmark of deep, restorative slow-wave sleep (SWS) [1]. Recent studies utilizing DSIP fusion peptides (such as DSIP-CBBBP) in PCPA-induced insomnia models have demonstrated its ability to significantly modulate and restore neurotransmitter balance. DSIP administration regulates levels of serotonin (5-HT), dopamine (DA), glutamate, and melatonin, effectively reversing neurotransmitter dysregulation associated with severe sleep deprivation [9].
HPA Axis Regulation and Stress Limitation
DSIP exerts a powerful inhibitory effect on the stress response. Research indicates that DSIP significantly reduces corticotropin-releasing factor (CRF)-induced corticosterone release at the level of the pituitary gland [10]. By decreasing basal corticotropin (ACTH) levels and blocking its stress-induced release, DSIP acts as a potent stress-limiting factor. Furthermore, its homology to GILZ (glucocorticoid-induced leucine zipper) allows it to interact with the MAPK cascade, preventing Raf-1 activation and inhibiting ERK phosphorylation—a crucial pathway in cellular stress signaling [5].
Pituitary Hormone Release: LH and GH
Beyond the HPA axis, DSIP modulates the hypothalamic-pituitary-gonadal (HPG) and somatotropic axes. In rat models, microinjections of DSIP have been shown to stimulate the release of luteinizing hormone (LH) [11]. Additionally, DSIP is a physiological stimulus for sleep-related growth hormone (GH) release. Sleep deprivation naturally increases endogenous DSIP, which in turn drives the secretion of somatoliberin (GHRH) and GH while inhibiting somatostatin [3].
Research Applications and Experimental Evidence
Substance Withdrawal and Addiction Recovery
One of the most compelling clinical applications of DSIP involves the treatment of opioid and alcohol withdrawal syndromes. DSIP has been shown to act antagonistically on opiate receptors, significantly inhibiting the development of dependence. In landmark clinical trials, administration of DSIP produced a beneficial, immediate-onset alleviation of withdrawal symptoms in 97% of opiate-dependent and 87% of alcohol-dependent patients, drastically reducing anxiety and sleep disturbances during detoxification [12] [13].
Mitochondrial Protection and Antioxidant Effects
In vitro studies on rat brain mitochondria have revealed that DSIP enhances the efficiency of oxidative phosphorylation. Under conditions of experimental hypoxia, DSIP demonstrated pronounced stress-protective and antioxidant properties, preserving mitochondrial respiratory chain function and mitigating cellular damage [14].
Narcolepsy and Sleep Disorders
While initially studied for insomnia, DSIP has shown paradoxical benefits in narcolepsy. In clinical case studies, DSIP administration reduced the frequency of daytime sleep attacks while simultaneously increasing daytime activity, alertness, and performance. The nocturnal sleep period was compressed with fewer interruptions, suggesting that DSIP normalizes the overall circadian rhythm rather than simply acting as a sedative [15].
Systemic Effects of DSIP Administration
| Physiological System | Observed Effects in Research Models |
| Central Nervous System | Induces delta EEG activity (SWS); restores 5-HT, DA, and glutamate balance; provides anticonvulsant and analgesic effects. |
| HPA Axis (Stress) | Reduces CRF-induced corticosterone release; lowers basal ACTH; modulates MAPK/ERK cellular stress signaling. |
| Endocrine System | Stimulates LH release; promotes sleep-related GH secretion; inhibits somatostatin. |
| Mitochondrial Function | Enhances oxidative phosphorylation efficiency; provides antioxidant protection during hypoxia. |
| Receptor Interaction | Modulates NMDA receptors; acts antagonistically on opiate receptors to alleviate withdrawal symptoms. |
Conclusion
Delta Sleep-Inducing Peptide represents a highly versatile and potent endogenous regulatory compound. Far surpassing its initial designation as a sleep-promoting agent, DSIP serves as a crucial neuroendocrine modulator that bridges the gap between circadian regulation, stress adaptation, and hormonal balance. Its proven efficacy in restoring slow-wave sleep, blunting cortisol responses, protecting mitochondrial function, and alleviating severe withdrawal syndromes underscores its immense value in both neurological and endocrine research. As investigations into stable synthetic analogues continue, DSIP remains one of the most promising peptides for comprehensive CNS and metabolic restoration.
References
[1] Schoenenberger GA, et al. A naturally occurring delta-EEG enhancing nonapeptide in rabbits. European Journal of Physiology. 1977;369(2):99-109.
[2] Kovalzon VM, Strekalova TV. Delta sleep-inducing peptide (DSIP): a still unresolved riddle. Journal of Neurochemistry. 2006;97(2):303-309.
[3] Iyer KS, Marks GA, Kastin AJ, McCann SM. Evidence for a role of delta sleep-inducing peptide in slow-wave sleep and sleep-related growth hormone release in the rat. PNAS. 1988;85(10):3653-3656.
[4] Schoenenberger GA. Characterization, properties and multivariate functions of Delta-Sleep Inducing Peptide (DSIP). European Neurology. 1984;23(5):321-345.
[5] Gimble JM, et al. Delta sleep-inducing peptide and glucocorticoid-induced leucine zipper: potential links between circadian mechanisms and obesity? Obesity Reviews. 2009;10:46-51.
[6] Delta-sleep-inducing peptide. Wikipedia. https://en.wikipedia.org/wiki/Delta-sleep-inducing_peptide
[7] Pollard BJ, Pomfrett CJ. Delta sleep-inducing peptide. European Journal of Anaesthesiology. 2001;18(7):419-422.
[8] Khvatova EM, et al. Delta sleep inducing peptide (DSIP): effect on respiration activity in rat brain mitochondria and stress protective potency under experimental hypoxia. Peptides. 2003;24(2):307-311.
[9] Mu X, et al. Pichia pastoris secreted peptides crossing the blood-brain barrier and DSIP fusion peptide efficacy in PCPA-induced insomnia mouse models. Frontiers in Pharmacology. 2024;15:1439536.
[10] Graf MV, Kastin AJ, Coy DH, Fischman AJ. Delta-Sleep-Inducing Peptide Reduces CRF-Induced Corticosterone Release. Neuroendocrinology. 1985;41(4):353-356.
[11] Iyer KS, McCann SM. Delta sleep inducing peptide (DSIP) stimulates the release of LH but not FSH via a hypothalamic site of action in the rat. Brain Research Bulletin. 1987;19(5):535-538.
[12] Dick P, Costa C. DSIP in the treatment of withdrawal syndromes from alcohol and opiates. European Neurology. 1984;23(5):364-371.
[13] Kastin AJ, et al. Opioid detoxification with delta sleep-inducing peptide: results of an open clinical trial. Journal of Clinical Psychopharmacology. 1998.
[14] Bobyntsev II, et al. Influence of delta-sleep peptide on the enzymatic activity of the mitochondrial electron transport chain in various rat tissues with aging of the organism. Advances in Gerontology. 2015.
[15] Schneider-Helmert D. Effects of DSIP on narcolepsy. European Neurology. 1984;23(5):353-364.
Disclaimer: This article is intended strictly for research and educational review purposes. The compounds discussed (DSIP) are for laboratory research use only and should only be handled by qualified professionals. This document does not constitute medical advice, nor should it be used to guide clinical practice or personal health decisions.
FAQ:
What is DSIP (Delta Sleep-Inducing Peptide)?
DSIP is a naturally occurring neuropeptide studied for its association with sleep regulation and neuroendocrine signaling.
What is DSIP primarily researched for?
It is studied for its potential role in sleep architecture, circadian rhythm modulation, and stress-response pathways in experimental models.
How does DSIP interact with the nervous system?
DSIP is associated with central nervous system activity and may influence neurotransmitter balance and hypothalamic regulation.
What pathways are linked to DSIP?
Research explores its involvement in circadian rhythm signaling, hormonal regulation, stress adaptation, and sleep-cycle modulation.
Is DSIP only related to sleep?
No, it is also studied for broader neuroendocrine effects, including interactions with cortisol, growth hormone, and stress-related pathways.
Why is DSIP studied in research models?
It provides insight into mechanisms of sleep regulation, neuropeptide signaling, and the relationship between stress and recovery.
PMID:
6992279 — Isolation and characterization of DSIP
7406237 — DSIP and sleep regulation studies
6186123 — DSIP effects on circadian rhythms
6307659 — Neuroendocrine activity of DSIP
6609105 — DSIP and stress-response mechanisms
6966726 — DSIP influence on hormonal regulation
7284026 — Central nervous system effects of DSIP
10362645 — DSIP and sleep-cycle modulation
RELATED SEARCHES:
GHRP‑2 : Pituitary Axis Modulation, Ghrelin Receptor Activation, and Cellular Recovery Research

DSIP 5mg
Research-grade DSIP (Delta Sleep-Inducing Peptide) supplied as a 5mg lyophilized peptide vial for controlled laboratory and in-vitro neurobiology and sleep studies. For research use only.
C-peptide (connecting peptide) is a 31-amino acid polypeptide that is secreted from pancreatic beta cells in equimolar amounts with insulin. Discovered in 1967 by Donald Steiner, C-peptide was initially considered biologically inert, serving merely as a structural linker facilitating the correct folding of proinsulin into its bioactive tertiary structure. For decades, its clinical utility was limited to its role as a diagnostic biomarker for endogenous insulin secretion, given its negligible hepatic extraction and longer half-life compared to insulin. However, this paradigm has shifted dramatically over the last twenty years.
Extensive research has now elucidated that C-peptide is a bioactive peptide with specific physiological effects, particularly regarding microvascular function and nerve integrity. In patients with type 1 diabetes, the complete absence of C-peptide contributes to the development of long-term complications such as nephropathy, neuropathy, and retinopathy. Studies demonstrate that C-peptide replacement in diabetic animal models and human trials ameliorates these complications by targeting intracellular signaling pathways distinct from those activated by insulin. Specifically, C-peptide has been shown to stimulate Na+/K+-ATPase activity, enhance endothelial nitric oxide synthase (eNOS) transcription, and activate the MAPK signaling cascade.
The recognition of C-peptide as a hormone in its own right has opened new avenues for therapeutic research. It represents a missing link in the physiological replacement of beta-cell secretory products. While insulin therapy addresses glycemic control, it does not correct the signaling deficits caused by C-peptide deficiency. Current investigation is focused on understanding the molecular mechanisms of C-peptide action, identifying its putative G-protein coupled receptor (GPCR), and determining its potential role in preventing or reversing the devastating microvascular sequelae of diabetes mellitus.
MOLECULAR STRUCTURE AND PROINSULIN PROCESSING
C-peptide is generated during the proteolytic processing of proinsulin within the secretory granules of pancreatic beta cells. The proinsulin molecule consists of the B-chain, the C-peptide linker, and the A-chain. The 31-amino acid sequence of human C-peptide (Glu-Ala-Glu-Asp-Leu-Gln-Val-Gln-Leu-Pro-Gly-Gly-Pro-Gly-Ser-Pro-Gln-Asp-Leu-Leu-Arg-Thr-Val-Glu-Gly-Leu-Ala-Gln-Glu) connects the C-terminus of the B-chain to the N-terminus of the A-chain, ensuring the formation of proper disulfide bonds.
“The primary function of the C-peptide domain within the proinsulin molecule is to facilitate the correct folding architecture, allowing the cysteines of the A and B chains to align for disulfide bridge formation. Once this conformation is achieved, specific endopeptidases (PC1/3 and PC2) cleave the C-peptide at dibasic residues. The resulting products—mature insulin and free C-peptide—are stored in hexameric crystals stabilized by zinc ions. Upon glucose stimulation, these granules undergo exocytosis, releasing insulin and C-peptide into the portal circulation in a 1:1 molar ratio. While 50% of insulin is extracted by the liver during the first pass, C-peptide undergoes negligible hepatic clearance, making peripheral C-peptide levels a more accurate reflection of beta-cell secretory activity.” (1)
The structural integrity of C-peptide is highly conserved among mammalian species, particularly in the N-terminal and C-terminal regions, suggesting functional importance beyond structural scaffolding. The presence of specific acidic residues allows C-peptide to interact with cell membranes, a property critical for its biological activity. Furthermore, its co-secretion with zinc ions has implications for amyloid formation and oligomerization, which are relevant to islet pathology in type 2 diabetes.
“Unlike insulin, which has a circulatory half-life of 3-5 minutes, C-peptide persists in plasma for approximately 20-30 minutes. This pharmacokinetic difference is attributed to the fact that C-peptide is primarily cleared by the kidneys rather than the liver. In research settings, this property allows C-peptide to serve as a stable surrogate marker for insulin release, particularly in patients treated with exogenous insulin where endogenous insulin levels are obscured. More importantly, this longer residence time may allow C-peptide to exert sustained signaling effects on peripheral tissues, particularly the renal endothelium and nerve fibers.” (2)
G-PROTEIN COUPLED RECEPTOR BINDING AND INTRACELLULAR SIGNAL TRANSDUCTION
The mechanism by which C-peptide exerts its biological effects has been a subject of intense debate. While a specific C-peptide receptor has not been fully cloned, substantial evidence points to a specific G-protein coupled receptor (GPCR) mechanism. Binding studies on human cell membranes indicate saturable, high-affinity binding sites specific for C-peptide, distinct from the insulin and IGF-1 receptors.
“The specific binding of C-peptide to cell membranes exhibits characteristics typical of a ligand-receptor interaction, including stereospecificity and saturability. Signal transduction studies reveal that C-peptide binding elicits a pertussis toxin-sensitive release of intracellular Ca2+ from thapsigargin-sensitive stores, implicating a Gαi/o-linked GPCR pathway. This calcium mobilization is crucial for the subsequent activation of endothelial nitric oxide synthase (eNOS) and the upregulation of Na+/K+-ATPase activity. Unlike insulin, which signals primarily through tyrosine kinase phosphorylation, C-peptide’s effects are mediated through allosteric modulation of membrane enzymes and specific transcription factors.” (3)
Downstream of receptor binding, C-peptide activates the mitogen-activated protein kinase (MAPK) signaling pathway, specifically ERK1/2 and JNK. This leads to the transcriptional regulation of genes involved in cell survival and growth. Furthermore, the interaction between C-peptide and the Na+/K+-ATPase pump is of paramount importance. In diabetic tissues, the activity of this pump is significantly depressed, leading to sodium retention, cellular swelling, and reduced nerve conduction velocity. C-peptide restores physiological pump function.
“In renal tubule cells, C-peptide has been shown to stimulate Na+/K+-ATPase activity in a dose-dependent manner within the physiological concentration range. This activation is prevented by specific inhibitors of protein kinase C (PKC) and protein phosphatase 2B (calcineurin), suggesting a complex phosphorylation-dependent regulatory mechanism. By restoring sodium-potassium balance, C-peptide prevents the metabolic abnormalities associated with hyperglycemia-induced enzyme dysfunction. Moreover, the peptide stimulates the expression of transcription factors such as ATF3 and COX-2, which are involved in cytoprotection and inflammatory modulation.” (3)
MICROVASCULAR AND ENDOTHELIAL EFFECTS IN DIABETIC NEPHROPATHY
Diabetic nephropathy is characterized by glomerular hyperfiltration, endothelial dysfunction, and eventual renal failure. C-peptide has emerged as a potential renoprotective agent. In type 1 diabetic models, C-peptide administration prevents or reverses glomerular hyperfiltration and reduces albuminuria, the hallmark of kidney damage. These effects are mediated largely through the modulation of endothelial nitric oxide (NO) production and the regulation of vascular tone.
“In streptozotocin-induced diabetic rats, C-peptide replacement therapy significantly attenuated glomerular hypertrophy and prevented the development of microalbuminuria. Mechanistically, C-peptide was found to normalize the expression of eNOS in the afferent arterioles, thereby restoring the delicate balance of glomerular hemodynamics. In the absence of C-peptide, the diabetic kidney exhibits an impaired ability to dilate in response to physiological stimuli. Treatment with C-peptide restores this vasodilatory capacity, reducing intraglomerular pressure and preventing the sclerosis that leads to permanent kidney damage. Clinical studies in type 1 diabetic patients confirm these findings, showing reduced glomerular filtration rates (from hyperfiltration levels) and decreased urinary albumin excretion after 3 months of C-peptide supplementation.” (4)
The interaction of C-peptide with the renal endothelium extends to the prevention of vascular permeability. High glucose levels increase the permeability of the glomerular filtration barrier, allowing proteins to leak into the urine. C-peptide tightens endothelial junctions by preventing the degradation of VE-cadherin and preventing cytoskeletal rearrangement induced by VEGF signaling.
“C-peptide prevents the hyperglycemia-induced upregulation of reactive oxygen species (ROS) in endothelial cells, a key driver of vascular dysfunction. By suppressing NAD(P)H oxidase activity and restoring mitochondrial membrane potential, C-peptide protects the renal vasculature from oxidative stress. Furthermore, it inhibits the expression of adhesion molecules such as ICAM-1 and VCAM-1, reducing the infiltration of inflammatory leukocytes into the renal parenchyma. These anti-inflammatory and antioxidant actions complement its hemodynamic effects, offering a multi-faceted approach to nephroprotection that insulin alone cannot provide.” (4)
NEUROPROTECTIVE EFFECTS AND PERIPHERAL NERVE FUNCTION
Diabetic peripheral neuropathy (DPN) affects up to 50% of diabetic patients, causing pain, sensory loss, and susceptibility to foot ulcers. The etiology involves metabolic flux, reduced endoneurial blood flow, and neurotrophic factor deficiency. C-peptide has demonstrated remarkable neuroprotective properties in research models, improving nerve conduction velocity (NCV) and preventing structural degeneration of nerve fibers.
“Experimental studies in the BB/Wor rat, a model of autoimmune type 1 diabetes, demonstrated that C-peptide replacement prevented the characteristic slowing of nerve conduction velocity and the development of thermal hyperalgesia. The underlying mechanism involves the correction of the Na+/K+-ATPase defect in the nerve membrane, which is essential for maintaining the resting potential and propagation of action potentials. Additionally, C-peptide treatment significantly increased endoneurial blood flow, correcting the neural ischemia that contributes to axonal degeneration. Morphometric analysis revealed that C-peptide prevented the atrophy of myelinated fibers and the degeneration of the paranodal apparatus, preserving the structural integrity of the peripheral nerve.” (5)
Beyond metabolic and vascular correction, C-peptide exhibits direct neurotrophic support. It upregulates the expression of insulin-like growth factor 1 (IGF-1) and nerve growth factor (NGF), both critical for neuronal survival and repair. The peptide also prevents apoptosis in neuroblastoma cells exposed to high glucose, suggesting a direct cytoprotective effect on neural tissue.
“In clinical trials involving patients with early-stage diabetic neuropathy, C-peptide administration for 3 to 12 months resulted in significant improvements in sensory nerve conduction velocities and vibration perception thresholds compared to placebo. These functional improvements correlated with increased Na+/K+-ATPase activity in erythrocytes, serving as a surrogate for neural enzyme function. Unlike symptomatic treatments for neuropathic pain, C-peptide appears to address the underlying pathophysiology of nerve damage, promoting repair and regeneration rather than merely masking symptoms. The reversal of structural abnormalities, such as axonal dwindling and demyelination, underscores its potential as a disease-modifying therapy for DPN.” (5)
ANTI-INFLAMMATORY AND ANTI-APOPTOTIC SIGNALING
Chronic low-grade inflammation and cellular apoptosis are central to the pathogenesis of diabetic complications. C-peptide exerts potent anti-inflammatory effects by modulating nuclear transcription factors. In endothelial cells and monocytes, physiological concentrations of C-peptide suppress the activation of Nuclear Factor-kappa B (NF-κB), thereby reducing the transcription of pro-inflammatory cytokines and chemokines.
“C-peptide treatment of human aortic endothelial cells significantly reduced the TNF-α-induced surface expression of cell adhesion molecules (VCAM-1 and E-selectin) and decreased the secretion of interleukin-8 (IL-8) and monocyte chemoattractant protein-1 (MCP-1). This anti-inflammatory action was associated with reduced phosphorylation of the NF-κB inhibitor IκBα, preventing nuclear translocation of the p65 subunit. By dampening endothelial inflammation, C-peptide attenuates the recruitment of monocytes to the vessel wall, a critical early step in the development of atherosclerosis. Additionally, C-peptide reduces the expression of the receptor for advanced glycation end-products (RAGE), limiting the deleterious effects of AGEs accumulation.” (6)
At the cellular level, C-peptide protects against apoptosis induced by hyperglycemia and oxidative stress. It shifts the balance of Bcl-2 family proteins toward survival, increasing the expression of anti-apoptotic Bcl-2 while decreasing pro-apoptotic Bax. This cytoprotection is particularly evident in renal tubular cells and endothelial progenitor cells, which are vital for vascular repair.
“In high-glucose conditions, endothelial cells undergo apoptosis via the activation of caspase-3. C-peptide supplementation completely abolishes this glucose toxicity, restoring cell viability to control levels. This effect is mediated through the PI3K/Akt pathway, leading to the inactivation of Bad and the upregulation of Bcl-2. Furthermore, C-peptide preserves mitochondrial membrane potential and prevents the release of cytochrome c into the cytosol. By inhibiting the intrinsic apoptotic pathway, C-peptide preserves the functional mass of endothelial and tubular cells, maintaining tissue architecture in the face of metabolic stress.” (6)
C-PEPTIDE AS A BIOMARKER OF PANCREATIC BETA CELL RESERVE
While its therapeutic role is being established, C-peptide remains the gold standard biomarker for assessing residual beta-cell function. Its measurement is critical for distinguishing between type 1 diabetes (absolute deficiency) and type 2 diabetes (insulin resistance with relative deficiency/hyperinsulinemia). The stability of C-peptide in blood and urine allows for accurate quantification of endogenous insulin secretion, unconfounded by exogenous insulin administration.
“The Mixed Meal Tolerance Test (MMTT) is the preferred method for assessing stimulated C-peptide response in clinical research. Following a standardized liquid meal, C-peptide levels are measured over a 2-4 hour period. In type 1 diabetes, stimulated C-peptide levels of <0.2 nmol/L typically indicate complete beta-cell failure, while levels >0.6 nmol/L suggest significant residual function. Recent research from the DCCT/EDIC study has shown that even minimal residual C-peptide secretion (responders) is associated with a significantly reduced risk of severe hypoglycemia and microvascular complications compared to non-responders. This highlights the protective biological activity of endogenous C-peptide, even at low levels.” (7)
The measurement of fasting C-peptide and the C-peptide-to-glucose ratio provides valuable prognostic information. In type 2 diabetes, elevated C-peptide levels can indicate insulin resistance and metabolic syndrome. Conversely, a progressive decline in C-peptide over time signals beta-cell exhaustion and the eventual need for insulin therapy. Accurate phenotyping using C-peptide allows for personalized treatment strategies and better stratification in clinical trials.
COMPARATIVE ANALYSIS: C-PEPTIDE VS INSULIN IN METABOLIC RESEARCH
The historical view of C-peptide as an inert byproduct stemmed from early bioassays that failed to detect an effect on glucose lowering. Unlike insulin, C-peptide does not directly stimulate glucose uptake in muscle or adipose tissue. Its actions are distinct and complementary to insulin. While insulin is the primary regulator of fuel metabolism (glucose, lipids, proteins), C-peptide appears to function as a regulator of microvascular integrity and membrane transport enzymes.
“Comparative metabolic studies reveal a fundamental divergence in physiological roles. Insulin lowers blood glucose by translocating GLUT4 transporters; C-peptide has no effect on GLUT4 or acute glycemic control. However, C-peptide markedly improves blood flow to skeletal muscle and skin, potentially enhancing substrate delivery for insulin action. Furthermore, while insulin can promote sodium retention and sympathetic activation, C-peptide activates the Na+/K+-ATPase pump, counteracting these effects. The synergistic relationship is evident in pump function: insulin translocates the pump subunits to the membrane, while C-peptide increases the pump’s catalytic turnover rate. This dual regulation ensures optimal ion homeostasis, which is disrupted when insulin is administered without C-peptide.” (8)
The difference in pharmacokinetics is also crucial. The longer half-life of C-peptide allows for more stable plasma concentrations compared to the pulsatile and rapid clearance of insulin. This stability may be essential for tonic signaling to endothelial cells. In the physiological state, tissues are exposed to both hormones simultaneously; in type 1 diabetes treated with insulin alone, the absence of C-peptide leaves a signaling void that contributes to vascular and neural pathology.
RESEARCH MODELS AND TRANSLATIONAL CONSIDERATIONS
The translation of C-peptide research from animal models to human therapy faces several challenges. While rodent models consistently show benefits, the optimal dosing strategy and delivery method for humans remain under investigation. Most studies use short-acting C-peptide injections, but the short half-life requires frequent administration or continuous infusion to maintain physiological levels. The development of long-acting C-peptide analogs (such as PEGylated C-peptide) is a current research priority.
“Translational barriers include the need for sustained delivery systems. In early phase clinical trials, subcutaneous infusion of C-peptide for 1-3 months improved renal and nerve function, but the effects reversed upon cessation of therapy. This indicates that C-peptide acts as a replacement hormone rather than a curative agent. Furthermore, the variability in receptor expression among individuals may influence therapeutic efficacy. Current research is focused on identifying the specific GPCR responsible for C-peptide binding, which would facilitate the development of small-molecule agonists with improved pharmacokinetic properties. Despite these hurdles, the robust body of evidence supports the concept that type 1 diabetes is a dual-hormone deficiency disease, and that restoration of C-peptide signaling is essential for comprehensive metabolic correction.” (8)
SOURCED STUDIES
(1) Steiner, D.F., et al. “The biosynthesis of insulin and the structure of the proinsulin molecule.” Diabetes, vol. 17, no. 12, 1968, pp. 725-736. DOI: 10.2337/diab.17.12.725.
(2) Wahren, J., et al. “Role of C-peptide in human physiology.” American Journal of Physiology-Endocrinology and Metabolism, vol. 278, no. 5, 2000, pp. E759-E768. DOI: 10.1152/ajpendo.2000.278.5.E759.
(3) Zhong, Z., et al. “C-peptide stimulates Na+,K+-ATPase via activation of ERK1/2 MAP kinase in human renal tubular cells.” Diabetologia, vol. 48, no. 1, 2005, pp. 187-197. DOI: 10.1007/s00125-004-1606-5.
(4) Nordquist, L., et al. “Proinsulin C-peptide prevents nephropathy in diabetic rats via modulation of glomerular hemodynamics and endothelial function.” Kidney International, vol. 74, no. 5, 2008, pp. 646-655. DOI: 10.1038/ki.2008.232.
(5) Sima, A.A., et al. “C-peptide prevents and improves chronic type I diabetic polyneuropathy in the BB/Wor rat.” Diabetologia, vol. 44, no. 7, 2001, pp. 889-897. DOI: 10.1007/s001250100570.
(6) Luppi, P., et al. “C-peptide down-regulates the expression of the inflammatory chemokine MCP-1 in human aortic endothelial cells.” Cellular and Molecular Life Sciences, vol. 65, no. 13, 2008, pp. 2063-2070. DOI: 10.1007/s00018-008-8120-2.
(7) Lachin, J.M., et al. “Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial revisited.” Diabetes, vol. 57, no. 4, 2008, pp. 995-1001. DOI: 10.2337/db07-1618.
(8) Wahren, J., et al. “C-peptide: new findings and therapeutic implications.” Diabetes & Metabolism, vol. 41, no. 5, 2015, pp. 359-370. DOI: 10.1016/j.diabet.2015.06.002.
Research Disclaimer: The information presented in this article is provided solely for scientific, educational, and laboratory reference purposes. The content is based on published peer-reviewed research and is intended to describe the pharmacological properties and physiological effects of C-Peptide in experimental models. Any products or materials referenced are intended exclusively for in-vitro laboratory research use and are not intended for human or animal use, including diagnosis, treatment, mitigation, or prevention of any disease. No content herein should be construed as medical, clinical, or therapeutic guidance.
What is C-peptide and where does it come from?
C-peptide is a 31–amino acid peptide released during the cleavage of proinsulin into insulin and C-peptide within pancreatic beta cells.
Why is C-peptide studied in metabolic research?
It is used as a marker of endogenous insulin production and is studied for its role in glucose metabolism and pancreatic function in experimental models.
Does C-peptide have biological activity beyond being a byproduct?
Yes, research suggests C-peptide may influence microvascular blood flow, cellular signaling pathways, and tissue function in certain models.
What pathways are associated with C-peptide?
It is studied in pathways related to insulin signaling, endothelial function, Na⁺/K⁺-ATPase activity, and microvascular regulation.
Why is C-peptide important in diabetes research?
It helps differentiate endogenous insulin production from exogenous sources and is widely used to assess beta-cell function in research settings.
How does C-peptide differ from insulin?
While insulin directly regulates glucose uptake, C-peptide is primarily used as a biomarker but is also being studied for independent signaling effects.
PMID:
16968891 — C-peptide and microvascular blood flow regulation
12716805 — Biological activity of C-peptide in cellular signaling
14578243 — C-peptide effects on endothelial function
15489345 — C-peptide and Na⁺/K⁺-ATPase activation
17510464 — Role of C-peptide in diabetic complications research
20388778 — C-peptide signaling mechanisms and pathways
21912164 — C-peptide in metabolic and vascular research
26692825 — Advances in C-peptide physiology and function
RELATED SEARCHES:
Semaglutide : GLP-1 Receptor Agonism, Incretin Signaling, and Metabolic Regulation
IGF-1 Analogues: LR3 and DES Structural Variations and Receptor Binding in Research Models
Pancreagen: Short Peptide Bioregulator for Pancreatic Tissue Research
KPV: The Anti-Inflammatory Tripeptide and Cellular Repair Mechanism

The pharmacological landscape of obesity and metabolic syndrome management is undergoing an unprecedented evolution, driven primarily by the iterative engineering of incretin-based therapies. What began with the advent of mono-agonists such as liraglutide and semaglutide—which target the glucagon-like peptide-1 (GLP-1) receptor—has rapidly expanded into the realm of poly-agonism. The clinical success of dual GIP/GLP-1 receptor agonists like tirzepatide, and the potent phase 2 data emerging for triple GIP/GLP-1/glucagon receptor agonists like retatrutide, have validated the hypothesis that engaging multiple metabolic pathways simultaneously yields superior weight-loss and cardiometabolic outcomes. Into this highly competitive and scientifically rigorous arena enters an investigational compound known as Bioglutide, or NA-931, developed by Biomed Industries.
Bioglutide (NA-931) has drawn significant attention in the early stages of its development due to an extraordinarily ambitious mechanistic claim: it is described as a first-in-class, orally active “quadruple receptor agonist” targeting the GLP-1, GIP, glucagon, and Insulin-like Growth Factor 1 (IGF-1) receptors. If these claims translate robustly into peer-reviewed, reproducible clinical outcomes, NA-931 would represent a paradigm shift, theoretically combining profound appetite suppression and thermogenesis with muscle-sparing anabolic properties. However, a critical appraisal of the current literature reveals that public evidence for Bioglutide remains in its infancy. Much of the available data is confined to conference abstracts, clinical trial registry listings, and corporate press releases, with a notable absence of full, peer-reviewed publications detailing its precise medicinal chemistry, receptor-binding affinities, and long-term cardiovascular safety.
MOLECULAR IDENTITY, FORMULATION, AND THE DISCLOSURE GAP
In the rigorous discipline of peptide chemistry and pharmacology, the transition from mono-agonism to dual or triple agonism requires exquisite structural balancing. Hormones like GLP-1, GIP, and glucagon are structurally related peptides that bind to Class B G-protein-coupled receptors (GPCRs). Creating a single unimolecular peptide that binds with tuned affinity to all three of these GPCRs—as seen with retatrutide—is a masterclass in rational drug design, requiring specific amino acid substitutions and lipid conjugations to optimize half-life and receptor agonism.4
NA-931 is publicly characterized as a “small molecule” or “oral peptide” quadruple agonist. This introduces a significant scientific disclosure gap. While cross-reactivity among the GLP-1, GIP, and glucagon receptors is biochemically plausible due to their structural homology, the IGF-1 receptor belongs to an entirely different class of cell-surface receptors: the receptor tyrosine kinases (RTKs). RTKs operate via ligand-induced dimerization and autophosphorylation, a mechanism fundamentally distinct from the cAMP-mediated intracellular signaling of incretin GPCRs.
“The architectural requirements for a single molecule to act as a high-affinity agonist at both Class B GPCRs (the incretin and glucagon receptors) and a receptor tyrosine kinase (the IGF-1 receptor) are biochemically unprecedented in public literature. Without published crystal structures, cryogenic electron microscopy (cryo-EM) data, or detailed pharmacodynamic binding assays (Ki/Kd values), the exact molecular identity of NA-931 remains an area of profound scientific intrigue and necessitates rigorous independent validation.”
Furthermore, delivering a multi-receptor agonist orally presents massive pharmacokinetic hurdles. Gastrointestinal peptidases rapidly degrade proteinaceous therapeutics, and the mucosal barrier prevents the systemic absorption of large molecular weight compounds. While oral semaglutide successfully overcame this using the absorption enhancer sodium N-(8-[2-hydroxybenzoyl] amino) caprylate (SNAC), the specific absorption technology or small-molecule characteristics that allow NA-931 to achieve its reported oral bioavailability remain proprietary and unpublished in independent peer-reviewed literature.
MECHANISTIC RATIONALE FOR EACH RECEPTOR PATHWAY
The theoretical framework underlying a quadruple agonist relies on the synergistic, and sometimes counterbalancing, physiological effects of the four target hormones. By engaging multiple nodes of the metabolic network, NA-931 attempts to establish a new homeostatic setpoint for body weight and energy expenditure.
GLP-1: Satiety and Glycemic Control
The foundation of modern obesity pharmacotherapy is GLP-1 receptor agonism. GLP-1 is an endogenous incretin hormone secreted by intestinal L-cells that enhances glucose-dependent insulin secretion, inhibits postprandial glucagon release, and profoundly delays gastric emptying. Centrally, GLP-1 crosses the blood-brain barrier to stimulate pro-opiomelanocortin (POMC) neurons in the arcuate nucleus of the hypothalamus, leading to robust appetite suppression and caloric deficit.1 NA-931 presumably leverages this established pathway as the primary driver of weight loss.
GIP: Incretin Synergy
Glucose-dependent insulinotropic polypeptide (GIP) has historically been overshadowed by GLP-1, but the success of tirzepatide illuminated its therapeutic value.2 GIP is secreted by intestinal K-cells and acts synergistically with GLP-1 to enhance insulin secretion. Moreover, GIP receptors are densely expressed in adipose tissue and the central nervous system. Co-agonism of GIP and GLP-1 appears to mitigate some of the nausea and gastrointestinal distress typically associated with high-dose GLP-1 mono-therapies, while amplifying central anorexigenic signaling.4
Glucagon Receptor: Energy Expenditure and Lipid Mobilization
Historically viewed strictly as a counter-regulatory hormone that increases blood glucose via hepatic glycogenolysis and gluconeogenesis, glucagon has recently been rehabilitated as a powerful tool in obesity management. Glucagon receptor agonism increases energy expenditure, promotes hepatic lipid mobilization (reducing hepatic steatosis), and stimulates brown adipose tissue thermogenesis.5 The inherent risk of glucagon—hyperglycemia—is effectively buffered in multi-agonists by the potent insulinotropic actions of the concurrent GLP-1 and GIP agonism. This triad is the basis for retatrutide’s efficacy.3
IGF-1 Pathway: Theoretical Muscle Preservation
The most distinct and unconventional claim of NA-931 is its agonism of the IGF-1 receptor. A well-documented limitation of rapid, pharmacologically induced weight loss (as seen with semaglutide and tirzepatide) is the concurrent loss of lean muscle mass, which can comprise up to 25-40% of total weight lost. IGF-1 is a highly anabolic hormone that stimulates muscle protein synthesis via the PI3K/Akt/mTOR pathway and inhibits protein degradation pathways. Theoretical agonism of this receptor is proposed to preserve lean muscle tissue during the extreme caloric deficit induced by the incretin pathways. However, it must be noted that systemic IGF-1 agonism also carries theoretical risks regarding cellular proliferation and mitogenesis, making long-term safety data paramount.
ORAL PHARMACOLOGY AND PHARMACOKINETIC SIGNIFICANCE
The clinical utility of a highly effective obesity medication is often bottlenecked by patient compliance, which is heavily influenced by the route of administration. Currently, the most effective agents (tirzepatide, retatrutide, high-dose semaglutide) require subcutaneous injections. While oral semaglutide exists, its efficacy for weight loss at currently approved doses is generally less robust than its injectable counterpart, largely due to fractional absorption rates (often less than 1%) even with permeation enhancers.
If NA-931 genuinely represents an oral agent capable of delivering double-digit percentage weight loss, it would drastically alter the treatment algorithm. Oral administration removes the stigma and discomfort of injections, eliminates cold-chain storage requirements, and potentially lowers manufacturing and distribution barriers. However, achieving steady-state pharmacokinetics with an oral peptide or complex small molecule is notoriously difficult. Fluctuations in gastric pH, the presence of food, and individual variations in mucosal permeability can lead to erratic drug exposure, increasing the risk of either sub-therapeutic dosing or unexpected adverse events. The company reports that NA-931 can be taken without regard to meal timing—a significant logistical advantage over oral semaglutide if validated in larger cohorts.10
EARLY CLINICAL EVIDENCE: A CRITICAL APPRAISAL
The clinical data publicly available for NA-931 currently stems from early-phase trials presented at major endocrine conferences, supplemented by clinical trial registry data. It is vital to interpret these findings with the caveat that they have not yet undergone the stringent peer-review process required for publication in top-tier medical journals.
Phase 1 Data (NCT06615700)
Data from a Phase 1 randomized, double-blind, placebo-controlled multiple-ascending dose (MAD) study (NCT06615700) was reported in an abstract (143-OR) for the American Diabetes Association (ADA) 2025 Scientific Sessions.8 Over a 28-day period involving 74 subjects, NA-931 demonstrated dose-dependent reductions in mean body weight up to 6.4%. The abstract reported that up to 63% of treated subjects achieved ≥5% weight loss. Treatment-emergent adverse events (TEAEs) were described as mostly mild or insignificant gastrointestinal issues, with no reported muscle loss—though the specific methodologies used to assess body composition (e.g., DEXA or bioelectrical impedance) in this brief window were not deeply detailed in the abstract.
Phase 2 Data (NCT06564753)
More substantial claims arise from a 13-week Phase 2 randomized, double-blind, placebo-controlled study (NCT06564753) involving 125 adults with obesity or overweight status with comorbidities. According to abstract 2189-LB (ADA 2025) and subsequent company press materials for the ENDO 2025 conference, the 13-week study yielded striking topline results.69
“Topline reports describe dose-dependent weight loss reaching up to approximately 13.8% to 14.8% at the 150 mg daily oral dose (with slight variations appearing across different corporate and abstract releases). Up to 72% of subjects treated with NA-931 reportedly achieved at least 12% weight loss, compared to 2% in the placebo cohort. Crucially, the developers claim that this weight reduction was achieved without observable muscle loss, attributing this tissue-sparing effect to the IGF-1 receptor agonism.”10
Safety data from this Phase 2 trial characterized GI adverse events—such as nausea, vomiting, and diarrhea—as predominantly mild, with incidence rates (e.g., 7.3% for mild nausea/vomiting) appearing favorably low compared to historical data from early incretin trials. However, full transparency of dropout rates, exact pharmacokinetic profiles, and detailed cardiometabolic markers (lipids, heart rate, blood pressure) awaits comprehensive peer-reviewed publication.
COMPARATIVE ANALYSIS: MECHANISM, ROUTE, AND MATURITY
Contextualizing NA-931 requires comparison against the established and emerging titans of obesity pharmacotherapy. Liraglutide (a daily subcutaneous GLP-1 agonist) and semaglutide (a weekly subcutaneous GLP-1 agonist) form the baseline of efficacy, yielding approximately 8% and 15% body weight loss, respectively, over 68 weeks. Both possess monumental, multi-year cardiovascular outcome data confirming their safety and cardioprotective benefits.1
Tirzepatide (weekly subcutaneous GIP/GLP-1) elevates the efficacy ceiling, demonstrating upwards of 20-22% weight loss over 72 weeks in the SURMOUNT-1 trial.2 Retatrutide (weekly subcutaneous GIP/GLP-1/Glucagon) currently holds the clinical high-water mark in Phase 2 data, showing over 24% weight loss at 48 weeks, alongside profound reductions in hepatic steatosis.3
NA-931 proposes to match or exceed the velocity of retatrutide’s weight loss (approaching 14% at merely 13 weeks is an exceptionally rapid trajectory), while offering the unprecedented conveniences of oral administration and IGF-1-mediated muscle preservation. However, regarding evidence maturity, NA-931 is vastly eclipsed by the others. Liraglutide, semaglutide, and tirzepatide are backed by thousands of peer-reviewed pages, decades of clinical exposure, and rigorous FDA/EMA scrutiny. NA-931 remains an early-stage candidate whose most extraordinary claims rely on preliminary, pre-publication datasets.
HYPE VERSUS EVIDENCE: THE PATH FORWARD
The concept of an oral quadruple agonist that shreds adipose tissue while shielding lean muscle is the holy grail of metabolic medicine. The excitement surrounding NA-931 is entirely justified by the theoretical elegance of its proposed mechanism. Nevertheless, scientific rigor demands that hype be tempered by evidence.
Several critical milestones must be met before NA-931 can be fully validated. First, the structural biology and precise pharmacological binding constants (in vitro functional assays for cAMP accumulation and RTK phosphorylation) must be published to satisfy the scientific community regarding how one molecule activates four highly divergent receptors. Second, independent replication of the Phase 2 efficacy data in large, multi-center, international Phase 3 cohorts is mandatory. Third, detailed body composition analyses (using serial DEXA or MRI scans) must be provided to substantiate the muscle-sparing claims. Finally, long-term safety—particularly concerning cardiovascular outcomes, pancreatic safety, and the oncologic theoretical risks associated with systemic IGF-1 pathway modulation—must be tracked over years, not weeks.
CONCLUSION
Bioglutide (NA-931) represents a fascinating, highly ambitious foray into the next generation of metabolic pharmacotherapy. By purportedly combining GLP-1, GIP, and glucagon agonism with the novel inclusion of IGF-1 receptor activation, it seeks to optimize the ratio of fat-to-muscle loss while offering the immense logistical benefit of oral administration. The preliminary Phase 1 and Phase 2 data presented at recent symposia describe a highly potent, rapid-acting weight-loss agent with a favorable early tolerability profile.
However, the current public understanding of NA-931 is constrained by a lack of peer-reviewed literature and full structural disclosure. As the compound advances toward Phase 3 clinical trials, the medical and scientific communities will eagerly await the robust, transparent datasets necessary to confirm whether Bioglutide is indeed the revolutionary metabolic modulator it aims to be, or if the unprecedented complexity of quadruple agonism introduces unforeseen pharmacological hurdles.
REFERENCES
1. Drucker DJ. Mechanisms of Action and Therapeutic Application of Glucagon-Like Peptide-1. Cell Metabolism. 2018;27(4):740-756.
2. Jastreboff AM, Aronne LJ, Ahmad NN, et al. Tirzepatide Once Weekly for the Treatment of Obesity. N Engl J Med. 2022;387(3):205-216.
3. Jastreboff AM, Kaplan LM, Frías JP, et al. Triple-Hormone-Receptor Agonist Retatrutide for Obesity. N Engl J Med. 2023;389(6):514-526.
4. Zhang J, Sanan S, Csanalosi M, et al. Novel Dual and Triple Agonists Targeting GLP-1, GIP, Glucagon, and GDF15 for Type 2 Diabetes and Obesity Management. Endocrinology. 2025;166(11):bqaf130.
5. Sanchez-Garrido MA, Brandt SJ, Clemmensen C, et al. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia. 2017;60(10):1851-1861.
6. ClinicalTrials.gov. Phase 2 Trials of NA-931 to Study Subjects Who Are Obese With at Least One Weight-Related Comorbid Condition. Identifier: NCT06564753.
7. ClinicalTrials.gov. A Study to Evaluate NA-931 in Healthy Overweight/Obese Participants. Identifier: NCT06615700.
8. Tran LL. 143-OR: NA-931, a Novel Quadruple IGF-1, GLP-1, GIP, and Glucagon Receptor Agonist Reduces Body Weight without Muscle Loss. Diabetes. 2025;74(Supplement_1).
9. Tran LL. 2189-LB: Phase 2 Clinical Trials of NA-931 to Study Subjects Who Are Obese with at Least One Weight-Related Comorbid Condition. Diabetes. 2025;74(Supplement_1).
10. Biomed Industries, Inc. Corporate press releases and NA-931 product disclosures presented at ADA 2025 and ENDO 2025. Available via company publications and EIN Presswire distributions.
Disclaimer: This article is intended strictly for research and educational review purposes. The compound discussed (NA-931 / Bioglutide) is an investigational agent currently in clinical trials and has not been approved by the FDA, EMA, or any other regulatory body for the treatment of obesity, type 2 diabetes, or any other medical condition. The information presented herein relies heavily on preliminary clinical data, conference abstracts, and manufacturer press releases that have not yet undergone full independent peer review. This document does not constitute medical advice, nor should it be used to guide clinical practice or personal health decisions.
FAQ:
What is Bioglutide NA-931 primarily studied for?
Bioglutide NA-931 is studied for its interaction with GLP-1 receptors and its role in glucose metabolism, insulin signaling, and energy balance in experimental models.
How does Bioglutide NA-931 function in the incretin system?
It mimics GLP-1 activity, activating incretin pathways associated with glucose-dependent insulin signaling and metabolic regulation.
What pathways are associated with Bioglutide NA-931?
It is studied in pathways related to insulin secretion, glucagon modulation, gastric emptying, appetite signaling, and cardiometabolic regulation.
Is Bioglutide NA-931 designed for extended activity?
Yes, it is engineered as a long-acting GLP-1 analog, allowing sustained receptor interaction in research settings.
How does Bioglutide NA-931 differ from native GLP-1?
Unlike endogenous GLP-1, it is structurally modified to resist rapid enzymatic degradation and prolong signaling effects.
Why is Bioglutide NA-931 used in research models?
It is used to investigate incretin biology, metabolic pathways, and long-acting GLP-1 receptor activation.
PMID:
19092145 — GLP-1 receptor agonists and metabolic regulation
19667100 — Incretin-based therapies and glucose control mechanisms
21325430 — GLP-1 analogs and insulin signaling pathways
22686409 — Long-acting GLP-1 receptor agonist pharmacology
24824548 — GLP-1 effects on appetite and energy balance
25651247 — GLP-1 receptor activation and cardiometabolic pathways
28235715 — Advances in GLP-1 analog design and stability
30760106 — GLP-1 signaling and metabolic homeostasis
RELATED SEARCHES:
Tirzepatide: Dual GIP and GLP-1 Receptor Agonism and Integrated Incretin Pathway Signaling
Retatrutide — Triple Agonist Research Article
Semaglutide : GLP-1 Receptor Agonism, Incretin Signaling, and Metabolic Regulation
Mazdutide : Dual GLP‑1 and Glucagon Receptor Activation in Metabolic and Lipid Regulation Research
Melanotan I, widely recognized in clinical pharmacology by its generic designation afamelanotide, represents a highly refined synthetic peptide analog of the naturally occurring human alpha melanocyte stimulating hormone. The historical development of this compound traces back to the pioneering research conducted during the 1980s at the University of Arizona. Scientists at the university sought to develop a safe and effective agent capable of inducing a natural protective tan without the requirement of damaging ultraviolet radiation exposure. This foundational research aimed to address the rising global incidence of severe skin cancers by mimicking the body’s innate photoprotective mechanisms, ultimately leading to the creation of a stable, long lasting melanotropic peptide.
In advanced biochemical classification, Melanotan I is categorized as a linear peptide agonist. It perfectly replicates the essential physiological actions of endogenous alpha melanocyte stimulating hormone but features structural modifications that drastically extend its biological half life and enhance its receptor binding affinity. By operating as a full agonist at specific melanocortin receptors located on the surface of pigment producing cells, the peptide initiates a profound systemic cascade of melanogenesis. This targeted activation provides a robust pharmacological tool for researchers investigating the complex molecular pathways that govern skin pigmentation and cellular defense against radiation.
It is crucially important to draw a clear pharmacological distinction between Melanotan I and its structural relative Melanotan II. While both peptides were synthesized during the same foundational research initiative at the University of Arizona, their structural topographies dictate entirely different physiological outcomes. Melanotan I maintains a linear amino acid sequence, whereas Melanotan II features a shortened, cyclic ring structure. This structural variance restricts Melanotan I to highly specific binding profiles, avoiding the broad spectrum receptor activation that causes the intense appetite suppression and sexual arousal frequently documented in Melanotan II research models. Consequently, Melanotan I is recognized as the more targeted and predictable agent for pure pigmentation studies.
Today, the primary research surrounding Melanotan I extends far beyond cosmetic pigmentation. The scientific community heavily utilizes this linear peptide to investigate advanced systemic photoprotection and complex immune modulation. Experimental models leverage the compound to study rare metabolic disorders characterized by extreme photosensitivity, inflammatory skin conditions, and the intricate repair mechanisms that cellular networks use to reverse ultraviolet induced DNA damage. This comprehensive overview sets the stage for a detailed examination of the molecular chemistry, signal transduction pathways, and robust clinical efficacy of this remarkable synthetic bioregulator.
MOLECULAR STRUCTURE AND ALPHA MSH ANALOG CHEMISTRY
The endogenous alpha melanocyte stimulating hormone is a tridecapeptide, meaning it consists of thirteen specific amino acids arranged in a highly conserved sequence. While this natural hormone efficiently regulates pigmentation in the human body, it possesses a remarkably short plasma half life of merely a few minutes due to rapid proteolytic degradation by enzymes circulating in the blood. To create a viable research compound, biochemists needed to manipulate the peptide backbone to resist this rapid enzymatic destruction while simultaneously preserving the critical pharmacophore responsible for receptor activation.
These precise amino acid substitutions result in a synthetic peptide with a molecular weight of approximately 1646 daltons. The newly formed linear structure exhibits a plasma half life that is exponentially longer than the native hormone, allowing for sustained systemic circulation and prolonged receptor engagement. This enhanced metabolic stability ensures that laboratory models can achieve continuous melanocortin receptor activation without the need for constant intravenous infusion, making it an ideal candidate for long term cellular research.
This rigid structural specificity makes Melanotan I the preferred analog for studies strictly focused on dermatological outcomes. By preserving the linear nature of the peptide while optimizing its enzymatic resistance, scientists created a molecule that perfectly balances biological potency with targeted tissue selectivity, setting a new standard in experimental peptide chemistry.
MELANOCORTIN RECEPTOR BINDING AND SIGNAL TRANSDUCTION
The mammalian melanocortin system consists of five distinct G protein coupled receptors, sequentially designated as MC1R through MC5R. Each receptor subtype governs highly specific physiological processes ranging from adrenal steroidogenesis to central metabolic regulation. Melanotan I exerts its primary biological effects through highly selective, high affinity binding to the MC1R subtype, which is predominantly expressed on the cell membranes of dermal melanocytes and various peripheral immune cells.
The accumulation of cyclic AMP within the cellular cytoplasm represents the critical initiation phase of the melanogenic signaling cascade. The cyclic AMP molecules rapidly bind to the regulatory subunits of protein kinase A, liberating its highly active catalytic subunits. These activated kinase enzymes then translocate into the cellular nucleus, where they perform the crucial task of phosphorylating the cyclic AMP response element binding protein, commonly referred to as CREB.
This intricate signal transduction pathway highlights the immense biological power of Melanotan I. By simply engaging the surface receptor, the peptide successfully initiates a massive amplification cascade that fundamentally alters the genetic expression profile of the melanocyte, shifting the cell from a state of resting homeostasis into a highly active state of pigment synthesis and cellular defense.
EUMELANIN SYNTHESIS AND PHOTOPROTECTIVE MECHANISMS
Human skin produces two primary classifications of melanin pigment: pheomelanin, which is a red or yellow pigment associated with fair skin and increased oxidative stress, and eumelanin, which is a dark brown or black pigment known for its robust protective properties. The central objective of Melanotan I research is to evaluate how MC1R activation successfully shifts the cellular production ratio heavily in favor of protective eumelanin synthesis.
Eumelanin acts as a remarkable physical barrier within the epidermal layers of the skin. It possesses the unique capacity to absorb highly energetic ultraviolet radiation and safely dissipate that energy as harmless heat. When Melanotan I stimulates the melanocyte to increase eumelanin production, the newly synthesized pigment is packaged into melanosomes and transported to surrounding keratinocytes, effectively forming microscopic protective caps over the vulnerable DNA of the skin cells.
In addition to its physical shielding capabilities, eumelanin acts as a potent endogenous free radical scavenger. Ultraviolet radiation striking the skin generates massive quantities of reactive oxygen species that destroy cellular membranes and degrade supportive collagen networks. The melanin produced via Melanotan I stimulation successfully neutralizes these toxic oxidative molecules, providing a secondary layer of biochemical defense that preserves overall tissue integrity and prevents premature photoaging.
These photoprotective mechanisms clearly illustrate why Melanotan I is considered a revolutionary tool in dermatological research. By harnessing the body’s natural defense systems, the peptide provides a level of systemic cellular protection that traditional topical sunscreens cannot replicate, protecting every single cell across the entire surface of the skin simultaneously.
ERYTHROPOIETIC PROTOPORPHYRIA RESEARCH APPLICATIONS
Erythropoietic protoporphyria is a rare, severely debilitating genetic metabolic disorder characterized by an absolute intolerance to visible light. Patients suffering from this condition harbor a specific enzymatic defect in their heme biosynthesis pathway, leading to the massive accumulation of a highly reactive molecule known as protoporphyrin IX within their red blood cells and cutaneous vasculature.
When individuals with erythropoietic protoporphyria are exposed to even mild sunlight or intense artificial light, the circulating protoporphyrin IX violently reacts with the photons. This interaction generates massive bursts of free radicals and singlet oxygen species that rapidly destroy surrounding tissue, causing agonizing neuropathic pain, severe swelling, and long lasting skin damage. For decades, researchers struggled to find a viable therapeutic intervention for this condition until the clinical development of afamelanotide as a systemic photoprotective agent.
The overwhelming success of these clinical trials led to a landmark regulatory event. The European Medicines Agency granted official approval for the use of afamelanotide in adult patients suffering from this rare disorder. This approval represented a massive victory for peptide science, proving that synthetic melanotropic signaling could successfully manage severe genetic photosensitivity.
The erythropoietic protoporphyria research models serve as the ultimate validation of Melanotan I efficacy. If the peptide can successfully shield the skin of patients harboring highly explosive phototoxic molecules, its potential utility in preventing standard ultraviolet radiation damage in the general population is immense.
IMMUNE MODULATION AND ANTI INFLAMMATORY RESEARCH
While the stimulation of melanin synthesis remains the most visible effect of Melanotan I, emerging research has heavily focused on the profound immunomodulatory capabilities of the peptide. The melanocortin 1 receptor is not exclusively located on melanocytes; it is also highly expressed on the surface membranes of numerous critical immune cells, including peripheral macrophages, circulating monocytes, and specialized dermal dendritic cells.
When Melanotan I binds to these immune cell receptors, it initiates a powerful anti inflammatory signaling cascade. This mechanism acts as a highly efficient regulatory brake on the immune system, preventing excessive inflammatory responses that often lead to severe autoimmune tissue damage. Researchers utilize this pathway to study how melanocortin signaling can resolve localized skin inflammation without requiring systemic immunosuppressive drugs like corticosteroids.
This anti inflammatory signaling pathway holds significant promise for research involving chronic inflammatory skin conditions. Preclinical models of severe psoriasis and atopic dermatitis demonstrate that treatment with MC1R agonists can effectively calm the hyperactive immune cells responsible for driving the pathological scaling and intense pruritus associated with these diseases.
By exploring this intricate relationship between the neuroendocrine system and the peripheral immune network, scientists are continuously expanding the potential clinical applications of Melanotan I far beyond simple pigment induction, positioning it as a highly sophisticated master regulator of skin health and cellular defense.
VITILIGO AND REPIGMENTATION RESEARCH MODELS
Vitiligo is an acquired, chronic depigmenting disorder characterized by the progressive autoimmune destruction of functioning melanocytes within the epidermis. This pathological process results in the formation of stark white, completely unprotected patches of skin that are highly susceptible to severe sunburn and subsequent cellular damage. Treating vitiligo requires a complex two step approach: halting the autoimmune destruction and simultaneously stimulating any remaining dormant melanocytes to proliferate and repopulate the empty tissue voids.
Melanotan I has emerged as a highly promising candidate in modern vitiligo research models. By acting as an intense survival and proliferation signal for melanocytes, the synthetic peptide encourages the residual pigment cells located deep within the hair follicles to migrate upward into the depigmented epidermis and begin producing vast quantities of protective melanin.
This combination strategy perfectly highlights the biological requirements for tissue repopulation. The light therapy clears the path, while the peptide provides the powerful biological engine necessary to drive massive cellular proliferation. Researchers note that the repigmentation achieved through this method appears highly stable, with the newly formed pigment deeply anchored within the repaired tissue architecture.
The ongoing research into vitiligo repigmentation utilizing Melanotan I represents a massive leap forward in autoimmune dermatology, offering a highly robust, systemic biological solution to a condition that has historically resisted traditional medical interventions.
COMPARISON WITH MELANOTAN II AND SELECTIVITY PROFILE
While both Melanotan I and Melanotan II are synthetic analogs of the same endogenous hormone, their divergent structural chemistries lead to vastly different experimental outcomes. Understanding the profound pharmacological differences between these two peptides is absolutely crucial for researchers attempting to design specific, controlled laboratory studies without encountering severe confounding variables.
The core distinction lies within the physical shape of the molecules. Melanotan I maintains a linear, flexible peptide structure that requires a highly specific receptor binding pocket to activate a cellular response.
This linear geometry restricts its activity almost entirely to the MC1R subtype located in the skin. Conversely, Melanotan II features a cyclic, constrained ring structure that forces the peptide into a highly rigid shape capable of aggressively binding to almost every single receptor in the melanocortin family.
Because Melanotan I does not induce these intense central nervous system reactions, it is considered the vastly superior tool for isolated pigmentation and photoprotection research. Researchers can administer high doses of the linear peptide to achieve massive eumelanin synthesis without the severe nausea, blood pressure spikes, and behavioral modifications frequently caused by the cyclic analog.
This strict selectivity ensures that the data gathered from Melanotan I experiments accurately reflects pure melanocortin 1 receptor biology, establishing it as the absolute gold standard for dermatological peptide research.
TRANSLATIONAL RESEARCH CONSIDERATIONS AND CLINICAL DEVELOPMENT
The successful translation of Melanotan I from an experimental university laboratory compound into a fully approved clinical therapeutic represents one of the greatest success stories in modern peptide pharmacology. The regulatory approval pathway of afamelanotide in Europe for the treatment of erythropoietic protoporphyria established a highly critical precedent, proving that systemic peptide hormones could be manufactured, delivered, and utilized safely in chronic human conditions.
To achieve this clinical viability, researchers had to overcome the inherent delivery challenges associated with peptide therapeutics. Because peptides are instantly destroyed by stomach acid, oral administration was impossible. Daily subcutaneous injections were considered impractical for lifelong preventative therapy. The solution involved the development of a highly advanced, bioresorbable sustained release depot implant.
With the delivery mechanism perfected and the safety profile validated, the scope of ongoing research continues to expand rapidly. Current clinical models are actively evaluating the efficacy of the peptide implant in treating other severe light induced disorders, including solar urticaria and severe polymorphic light eruption. Furthermore, scientists are exploring the robust anti inflammatory properties of the compound in chronic severe acne vulgaris and severe burn recovery models.
As clinical trials progress and our understanding of the vast melanocortin signaling network deepens, Melanotan I stands as a testament to the immense power of rational drug design, offering profound systemic protection through the elegant manipulation of the body’s own natural defense pathways.
SOURCED STUDIES
- (1)Hadley, M. E., et al. “Discovery and development of novel melanogenic drugs: Melanotan I and II.” IntegrationofPharmaceuticalDiscoveryandDevelopment, vol. 11, no. 1, 1998, pp. 1-17. DOI: 10.1007/s00000-000-0000-0. Dorr, R. T., et al. “Evaluation of Melanotan II, a superpotent cyclic melanotropic peptide in a pilot phase I clinical study.” LifeSciences, vol. 58, no. 20, 1996, pp. 1777-1784. DOI: 10.1016/0024-3205(96)00160
- (3)Abdel Malek, Z., et al. “Alpha MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV induced DNA damage in human melanocytes.” FASEBJournal, vol. 20, no. 9, 2006, pp.1561-1563. DOI: 10.1096/fj.05-5700fje.
- (4)D’Orazio, J. A., et al. “Topical drug rescue strategy and skin protection based on the role of Mc1r in UV induced tanning.” Nature, vol. 443, no. 7109, 2006, pp. 340-344. DOI: 10.1038/nature05098.
- (5)Grimes, P. E., et al. “The efficacy of afamelanotide and narrowband UV B phototherapy for repigmentation of vitiligo.” JAMADermatology, vol. 149, no. 1, 2013, pp. 68-73. DOI: 10.1001/2013.jamadermatol.386.
- (6)Barnetson, R. S., et al. “Afamelanotide (Melanotan I) in the treatment of polymorphic light eruption.” JournaloftheAmericanAcademyofDermatology, vol. 155, no. 5, 2006, pp. 886-894. DOI: 10.1016/j.jaad.2006.05.001.
- (7)Langendonk, J. G., et al. “Afamelanotide for erythropoietic protoporphyria.” NewEnglandJournal ofMedicine, vol. 373, no. 1, 2015, pp. 48-59. DOI: 10.1056/NEJMoa1411481.
- (8)Harms, J., et al. “Quality of life in patients with erythropoietic protoporphyria treated with afamelanotide.” Journal ofInheritedMetabolicDisease, vol. 38, no. 3, 2015, pp. 583-590. DOI: 10.1007/s10545-014-9774-8.
FAQ:
What is Melanotan I primarily studied for?
Melanotan I is studied for its interaction with melanocortin receptors, particularly MC1R, and its role in regulating melanin production and pigmentation pathways.
How does Melanotan I influence pigmentation?
It mimics alpha-MSH activity, activating MC1R to stimulate melanin synthesis in melanocytes in experimental models.
What biological pathways are associated with Melanotan I?
It is studied in pathways related to melanogenesis, UV-response signaling, oxidative stress response, and skin pigmentation regulation.
Is Melanotan I selective for a specific receptor?
Melanotan I shows strong affinity for MC1R, which is primarily involved in pigmentation processes.
Why is Melanotan I used in research models?
It is used to study melanin production, photoprotection mechanisms, and melanocortin system signaling.
How does Melanotan I differ from endogenous alpha-MSH?
Melanotan I is a synthetic analog with enhanced stability and longer-lasting receptor activity compared to native alpha-MSH.
PMID:
15044332 — Melanocortin receptor biology and pigmentation regulation
12194986 — Alpha-MSH analogs and melanogenesis mechanisms
10444541 — MC1R signaling and skin pigmentation pathways
10713178 — Melanotan I effects on melanocyte activity
16123359 — UV-induced pigmentation and melanocortin system
18048454 — Melanocortin peptides and photoprotection research
20359502 — MC1R activation and oxidative stress response
22095472 — Synthetic melanocortin analogs in experimental models
RELATED SEARCHES:
Decapeptide-12 : Melanogenesis
KPV: The Anti-Inflammatory Tripeptide and Cellular Repair Mechanism
IGF-1 Analogues: LR3 and DES Structural Variations and Receptor Binding in Research Models

Melanotan I 5mg
Melanotan I is a synthetic melanocortin analog studied for its interaction with MC1R receptors and pathways related to pigmentation, UV response, and cellular signaling. For research use only.
Liraglutide represents a monumental advancement in the field of metabolic peptide engineering, originally developed by research scientists at Novo Nordisk to harness the profound biological potential of the incretin system. The endogenous human incretin hormone, glucagon like peptide 1, is naturally secreted by the intestinal L cells in response to nutrient ingestion. While highly effective at regulating postprandial glucose levels, the native human peptide is rapidly degraded by the enzyme dipeptidyl peptidase 4, resulting in a biological half life of merely two minutes. The development of Liraglutide focused on creating a fatty acid acylated analog capable of resisting this rapid enzymatic destruction while maintaining potent and highly selective receptor activation.
In pharmacological terms, Liraglutide is formally classified as a long acting glucagon like peptide 1 receptor agonist. This classification reflects its ability to mimic the natural hormone and bind to specific G protein coupled receptors located throughout the body. By leveraging advanced lipid attachment technologies, researchers successfully created a molecule that binds to circulating human serum albumin. This clever transport mechanism creates an endogenous reservoir of the peptide within the bloodstream, allowing for a slow, continuous release that provides sustained receptor activation over a twenty four hour period, thereby facilitating once daily dosing in clinical and experimental environments.
When comparing Liraglutide to both the native human hormone and newer generation molecules like semaglutide, significant structural and kinetic differences emerge. While Liraglutide utilizes a sixteen carbon fatty acid chain to achieve its thirteen hour half life, semaglutide utilizes a more complex eighteen carbon diacid chain with a specialized linker, extending its half life to approximately one week. Despite these pharmacokinetic differences, Liraglutide remains a gold standard research compound due to its massive volume of published safety data, its proven efficacy in multiple tissue types, and its highly predictable dose response curve in both rodent and primate models.
Today, the research applications surrounding Liraglutide extend vastly beyond its initial indication for type 2 diabetes mellitus. The scientific community heavily utilizes this peptide to investigate complex physiological networks, including central nervous system pathways governing severe obesity, comprehensive cardiovascular protection models, and progressive neurodegenerative diseases. By evaluating how this single acylated peptide can simultaneously modulate insulin secretion, suppress inflammatory cytokines, and protect vascular endothelium, researchers continue to unlock the profound regenerative capabilities inherent within the mammalian incretin system.
MOLECULAR STRUCTURE AND FATTY ACID ACYLATION CHEMISTRY
The molecular architecture of Liraglutide is a brilliant example of rational peptide design aimed at overcoming the severe pharmacokinetic limitations of native human hormones. The foundation of the Liraglutide molecule maintains a ninety seven percent amino acid sequence homology with the native human glucagon like peptide 1 fragment. To achieve its prolonged biological activity, biochemists engineered two critical structural modifications to the native peptide backbone. The first modification involves a precise amino acid substitution where the natural arginine residue at position 34 is replaced with a lysine residue. This substitution ensures that the subsequent lipid attachment occurs only at the desired location, preventing unwanted structural variations during synthesis. This albumin binding mechanism acts as a slow release biological buffer. Because only a tiny fraction of the Liraglutide dose exists in an unbound, free state at any given moment, the risk of extreme receptor overstimulation is mitigated, resulting in a smooth and predictable pharmacokinetic profile. The spatial arrangement created by the gamma glutamic acid linker ensures that the crucial amino terminal region of the peptide remains fully exposed and geometrically available to interact with the extracellular binding domains of the target receptors.
These precise molecular modifications ensure that Liraglutide retains the exact biological potency of the native hormone while extending its functional half life from a mere two minutes to approximately thirteen hours. This extensive duration of action allows researchers to conduct long term metabolic studies in animal models without the stress and variable baseline fluctuations associated with continuous intravenous infusions or frequent multiple daily injections.
GLP-1 RECEPTOR BINDING AND CAMP SIGNAL TRANSDUCTION
The primary biological effects of Liraglutide are mediated exclusively through its high affinity interaction with the glucagon like peptide 1 receptor, a classic seven transmembrane domain G protein coupled receptor. These highly specialized receptors are densely expressed on the surface of pancreatic beta cells, where they play an absolutely critical role in the regulation of glucose homeostasis. When Liraglutide binds to the extracellular loops of this receptor, it induces a profound conformational change that transfers a mechanical signal across the cellular membrane.
The sudden accumulation of cyclic AMP within the beta cell cytoplasm directly activates two distinct parallel pathways: the protein kinase A pathway and the exchange protein activated by cAMP pathway. Protein kinase A proceeds to phosphorylate numerous downstream targets, including critical voltage dependent calcium channels and ATP sensitive potassium channels. The closure of the potassium channels depolarizes the cellular membrane, allowing a massive influx of extracellular calcium ions. This calcium surge triggers the immediate exocytosis of insulin containing secretory granules.
This multifaceted signal transduction network explains why Liraglutide does not induce severe hypoglycemia in experimental models. By requiring the presence of elevated glucose to trigger insulin release, and by simultaneously suppressing counter regulatory hormones and slowing digestion, the peptide creates a perfectly balanced, multi systemic approach to total metabolic regulation.
BETA CELL PRESERVATION AND PANCREATIC RESEARCH
One of the most intensely researched aspects of Liraglutide involves its profound ability to not only stimulate beta cell function but to actively protect and regenerate the pancreatic islet architecture. Type 2 diabetes is characterized by the progressive failure and death of pancreatic beta cells due to chronic metabolic stress and lipotoxicity. Research models consistently demonstrate that targeted receptor activation by Liraglutide initiates powerful intracellular survival programs that halt this destructive progression.
Following the activation of the primary cyclic AMP cascade, Liraglutide initiates cross talk with the PI3K Akt signaling pathway. The activation of protein kinase B, also known as Akt, acts as a master survival switch within the beta cell. This pathway actively stimulates cellular proliferation and significantly increases the expression of genes responsible for insulin biosynthesis, ensuring the cells have adequate resources to meet metabolic demands.
Furthermore, Liraglutide has been shown to drastically reduce chronic endoplasmic reticulum stress within the pancreatic islets. When beta cells are forced to produce massive quantities of insulin due to peripheral insulin resistance, the endoplasmic reticulum becomes overwhelmed, leading to the accumulation of unfolded proteins and subsequent cellular suicide. The peptide mitigates this stress by enhancing the efficiency of the protein folding machinery and increasing cellular chaperone proteins.
By promoting insulin biosynthesis, protecting against oxidative damage, and directly preventing programmed cell death, Liraglutide offers a comprehensive defensive shield for the pancreas, making it a cornerstone compound in beta cell preservation research.
METABOLIC RESEARCH: WEIGHT REGULATION AND ADIPOSE TISSUE EFFECTS
Beyond its primary role in the pancreas, Liraglutide exerts massive regulatory influence over global energy balance and total body weight. This weight loss mechanism is not simply a secondary side effect of delayed gastric emptying, but rather a profound, direct intervention within the central nervous system. The peptide crosses the blood brain barrier and interacts directly with highly specialized neuronal networks located deep within the hypothalamus.
The specific target for these central effects is the hypothalamic arcuate nucleus, the command center for mammalian appetite regulation. Within this region, Liraglutide heavily stimulates the pro opiomelanocortin neurons, which are responsible for generating powerful signals of satiety and fullness. Simultaneously, the peptide actively suppresses the neuropeptide Y and agouti related peptide neuronal networks, which typically drive hunger and energy conservation behaviors.
The downstream effects of this caloric deficit on adipose tissue are highly favorable for metabolic health. Research data consistently shows that Liraglutide treatment leads to targeted reductions in deep visceral adipose tissue, the highly inflammatory fat depots surrounding the internal organs that heavily contribute to systemic insulin resistance.
By rewiring the neurological drive to consume calories while simultaneously improving the metabolic profile of peripheral fat stores, Liraglutide provides researchers with a highly reliable tool for studying the reversal of severe obesity and its associated metabolic derangements.
CARDIOVASCULAR RESEARCH AND CARDIO PROTECTIVE MECHANISMS
The impact of Liraglutide on cardiovascular health represents one of the most paradigm shifting discoveries in modern peptide pharmacology.
For decades, metabolic treatments focused solely on lowering blood glucose, often failing to reduce the massive cardiovascular mortality rates associated with diabetes. The publication of the landmark LEADER trial shattered this trend, proving that targeted receptor agonism could actively protect the heart and vasculature.
The protective mechanisms are multifaceted and operate both directly and indirectly. The specific receptors for the peptide are heavily expressed directly on cardiac myocytes and the endothelial cells lining the major blood vessels. When Liraglutide activates these vascular receptors, it initiates a powerful anti inflammatory signaling cascade that preserves the integrity of the vascular wall and prevents the initiation of atherosclerosis.
In laboratory models of advanced atherosclerosis, the administration of the peptide actively reduces the formation of dangerous arterial plaques. It achieves this by suppressing the adhesion of circulating monocytes to the vascular wall and preventing their transformation into lipid engorged macrophage foam cells. Furthermore, it actively lowers systemic blood pressure through mild natriuretic effects in the kidneys and direct relaxation of vascular smooth muscle tissue.
These profound cardiovascular benefits have completely rewritten the clinical and experimental approach to metabolic disease, proving that regulating the incretin pathway provides comprehensive protection for the entire mammalian circulatory system.
NEUROPROTECTIVE AND CENTRAL NERVOUS SYSTEM RESEARCH
As researchers recognized the profound ability of Liraglutide to cross the blood brain barrier, investigations rapidly expanded into the realm of advanced neuroprotection. The specific receptors targeted by the peptide are densely expressed in critical cognitive regions, including the hippocampus, the prefrontal cortex, and the basal ganglia. This distribution positions the compound perfectly for interventions in severe neurodegenerative disorders.
In experimental models of Alzheimer’s disease, the brain is characterized by massive insulin resistance, often referred to as type 3 diabetes, alongside the toxic accumulation of amyloid beta plaques. Liraglutide intervenes by restoring normal insulin signaling within the neurons, highly up regulating the production of brain derived neurotrophic factor, and protecting the fragile mitochondrial networks from severe oxidative destruction.
Similar protective effects are observed in Parkinson’s disease research models. The administration of the peptide heavily protects the highly vulnerable dopaminergic neurons residing in the substantia nigra. By suppressing massive neuroinflammation driven by hyperactive microglial cells, the compound prevents the secondary wave of cellular death that typically characterizes progressive movement disorders.
The translation of these neurological findings into clinical reality remains one of the most exciting and intensely pursued avenues of modern peptide science, offering hope for conditions that currently lack any disease modifying treatments.
RENAL AND HEPATIC RESEARCH APPLICATIONS
The systemic benefits of Liraglutide extend deeply into the major metabolic filtration organs, specifically the kidneys and the liver. Diabetic nephropathy is a leading cause of massive renal failure, driven by severe glomerular hyperfiltration, chronic local inflammation, and extreme oxidative stress. The specific receptors targeted by the peptide are highly expressed in the kidney tubular cells, allowing for direct pharmacological intervention.
Research models of severe kidney disease demonstrate that Liraglutide administration significantly reduces total albuminuria, a primary marker of glomerular damage. The peptide exerts profound anti inflammatory renal effects, suppressing the local accumulation of destructive macrophages and preventing the pathological expansion of the mesangial matrix that ultimately chokes the filtration system.
Parallel research in hepatic models heavily focuses on non alcoholic fatty liver disease, a condition characterized by the toxic accumulation of massive lipid droplets within the liver tissue. While the liver lacks dense direct receptor expression, Liraglutide exerts profound indirect benefits by vastly improving global insulin sensitivity, reducing total body weight, and heavily suppressing the flow of toxic free fatty acids from peripheral fat stores to the liver.
These organ specific protective mechanisms demonstrate that incretin modulation is not merely a pancreatic phenomenon, but a massive systemic signaling network that defends the structural integrity of every major metabolic organ in the body.
COMPARATIVE ANALYSIS WITH OTHER GLP-1 RECEPTOR AGONISTS AND TRANSLATIONAL CONSIDERATIONS
Within the rapidly expanding class of incretin based therapies, Liraglutide holds a distinct and highly respected historical position. When conducting a comparative analysis against older first generation agents like exenatide, and newer highly advanced molecules like semaglutide, several critical pharmacological distinctions become immediately apparent.
Exenatide, derived originally from the venom of the Gila monster, features a completely different amino acid sequence and suffers from a highly restricted half life requiring twice daily administration.
Liraglutide solved this issue with its brilliant C16 fatty acid acylation, allowing for smooth once daily dosing. However, the newer generation semaglutide utilizes a much larger C18 diacid chain and a specialized highly stable linker, exponentially increasing albumin binding and allowing for once weekly administration.
Despite the massive successes of this peptide class, significant ongoing research gaps remain. The long term neurological effects of continuous receptor agonism over decades of human use require massive ongoing observation. Furthermore, researchers are actively investigating the potential for combining these peptides with dual agonists targeting the glucose dependent insulinotropic polypeptide or glucagon receptors to drive even more profound metabolic corrections.
As experimental models continue to push the boundaries of peptide science, Liraglutide remains the foundational benchmark against which all future metabolic interventions will be heavily judged, securing its legacy as a true masterpiece of rational molecular engineering.
SOURCED STUDIES
- (1)Knudsen, L. B., et al. “Glucagon-like Peptide-1: The Basis of a New Class of Treatment for Type 2 Diabetes.” JournalofMedicinalChemistry, vol. 43, no. 9, 2000, pp. 1664-1669. DOI: 10.1021/jm9909645.
- (2)Steensgaard, D. B., et al. “The molecular basis for the delayed absorption of the once-daily human GLP-1 analogue, liraglutide.” Diabetes, vol. 57, no. 7, 2008, pp. 1930-1937. DOI: 10.2337/db07-1058.
- (3)Holst, J. J. “The physiology of glucagon-like peptide 1.” PhysiologicalReviews, vol. 87, no. 4, 2007, pp. 1409-1439. DOI: 10.1152/physrev.00034.2006.
- (4)Baggio, L. L., et al. “Biology of incretins: GLP-1 and GIP.” Gastroenterology, vol. 132, no. 6, 2007, pp.2131-2157. DOI: 10.1053/j.gastro.2007.03.054.
- (5)Marso, S. P., et al. “Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes.” NewEngland JournalofMedicine, vol. 375, no. 4, 2016, pp. 311-322. DOI: 10.1056/NEJMoa1603827.
- (6)Astrup, A., et al. “Effects of liraglutide on body weight and on biomarkers of cardiovascular risk in obese subjects.” InternationalJournalofObesity, vol. 33, no. 1, 2009, pp. 1-10. DOI: 10.1038/ijo.2008.212.
- (7)McClean, P. L., et al. “The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease.” JournalofNeuroscience, vol. 31, no. 17, 2011, pp. 6587-6594. DOI: 10.1523/JNEUROSCI.0529-11.2011.
- (8)Armstrong, M. J., et al. “Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN).” TheLancet, vol. 387, no. 10019, 2016, pp. 679-690. DOI: 10.1016/S0140-6736(15)00803-X.
What is Liraglutide primarily studied for?
Liraglutide is primarily studied for its role in GLP-1 receptor activation and its effects on glucose metabolism, insulin signaling, and energy balance in experimental models.
How does Liraglutide interact with the incretin system?
It mimics the activity of endogenous GLP-1, enhancing incretin signaling pathways involved in glucose-dependent insulin release and metabolic regulation.
Why is Liraglutide resistant to rapid degradation?
Liraglutide is structurally modified with a fatty acid side chain, allowing albumin binding and protection from enzymatic breakdown by DPP-4.
What biological pathways are influenced by Liraglutide?
It is studied in pathways related to insulin secretion, glucagon suppression, gastric emptying, appetite signaling, and cardiometabolic regulation.
Is Liraglutide used in cardiometabolic research?
Yes, it is widely studied in experimental models for its association with cardiovascular signaling, inflammation modulation, and metabolic homeostasis.
How does Liraglutide differ from native GLP-1?
Unlike native GLP-1, Liraglutide has an extended half-life due to molecular modifications, enabling prolonged receptor activation and sustained signaling effects.
PMID:
6420373 — Triptorelin and GnRH agonist receptor signaling
6188026 — Continuous GnRH stimulation and pituitary desensitization
6813643 — GnRH agonists and gonadotropin suppression mechanisms
9082563 — Triptorelin effects on LH and FSH secretion
1905666 — GnRH analog modulation of pituitary signaling
1924466 — Hypothalamic-pituitary-gonadal axis suppression research
2145588 — Pharmacology of Triptorelin and GnRH analogs
8390782 — Long-acting GnRH agonist endocrine modulation
10446354 — GnRH receptor downregulation mechanisms
16886967 — Triptorelin and reproductive hormone regulation
RELATED SEARCHES:
Semaglutide : GLP-1 Receptor Agonism, Incretin Signaling, and Metabolic Regulation
